Biosynthesis of polyketide synthase extender units
Yolande A. Chanab, Angela M. Podevelsa, Brian M. Kevanya and Michael G. Thomas*ab
aDepartment of Bacteriology, University of Wisconsin-Madison, Madison, WI 53706, USA. E-mail: thomas@bact.wisc.edu
bMicrobiology Doctoral Training Program, University of Wisconsin, Madison, WI 53706, USA
First published on 27th October 2008
Abstract
Covering: 1969 up to August 2008
This review covers the biosynthesis of extender units that are utilized for the assembly of polyketides by polyketide synthases. The metabolic origins of each of the currently known polyketide synthase extender units are covered.
Yolande A. Chan received her B.A. in Biology from Barnard College in 2000. In 2001, she began her graduate studies in microbiology at the Ohio State University and joined the laboratory of Prof. Michelle Rondon, studying nodulation competitiveness in rhizobia. She transferred to the University of Wisconsin–Madison in 2002 and joined the laboratory of Prof. Michael Thomas, where she studies unusual precursors in antibiotic biosynthesis. |
Angela M. Podevels received her B.S. in bacteriology from the University of Wisconsin–Madison in 2004. During her undergraduate years, with Prof. Timothy Donohue, she studied photosynthesis in Rhodobacter sphaeroides. She joined the laboratory of Prof. Thomas in 2004 and is studying various aspects of antibiotic biosynthesis. |
Brian M. Kevany received a B.S. in horticulture in 2002 from Michigan State University. He received his Ph.D. in 2007 in plant molecular and cellular biology in the laboratory of Prof. Harry Klee, focusing on the role of the multigene ethylene receptor family in tomato fruit development. He is currently a postdoctoral associate for Prof. Michael Thomas in the Department of Bacteriology at the University of Wisconsin–Madison. The focus of his research is in understanding the basic enzymology of polyketide and nonribosomal peptide synthases for use in novel antibiotic generation. |
Michael G. Thomas received a B.S. in biochemistry at Rutgers University in 1992. He received an M.S. in microbiology from Washington State University in 1994 under Prof. Kathleen Postle, focusing on the analysis of the TonB-dependent transport system involved in coenzyme B12, siderophore, and colicin uptake in E. coli. He received his Ph.D. in bacteriology from the University of Wisconsin–Madison in 1999 under the guidance of Prof. Jorge C. Escalante-Semerena, focusing on the biosynthesis of coenzyme B12 in bacteria and archaea. Michael was an NIH postdoctoral fellow with Prof. Christopher T. Walsh at Harvard Medical School, where he studied nonproteinogenic amino acid and nonribosomal peptide biosynthesis. In 2002 he started his own research group at the University of Wisconsin–Madison. In 2006 he was named the Alfred Toepfer Faculty Fellow in the College of Agriculture and Life Sciences at the University of Wisconsin–Madison. His research interests include natural product biosynthesis, discovery, and metabolic engineering. |
 From left to right: Brian M. Kevany, Angela M. Podevels, Yolande A. Chan and Michael G. Thomas |
1 Introduction
Polyketides are a large class of structurally diverse natural products exhibiting a vast array of biological and pharmacological activities such as antibacterial, antifungal, anticholesterol, antiparasitic, anticancer, and immunosuppressive properties. While structurally diverse, all polyketides are assembled by successive rounds of decarboxylative Claisen condensations between a thioesterified malonate derivative and an acyl thioester (Fig. 1). The enzymes that catalyze these condensations are referred to as polyketide synthases (PKSs). Because of the mechanistic similarities between fatty acid and polyketide biosynthesis, PKSs are classified using nomenclature for types of fatty acid synthases, with some modifications added as needed. There have been an extensive number of reviews on the enzymes associated with each type of PKS.1–5 Here we briefly summarize each type to set the stage for a more detailed discussion of the different types of PKS extender units, their biosynthesis, and their occurrence in different types of PKSs with an emphasis on bacterial systems.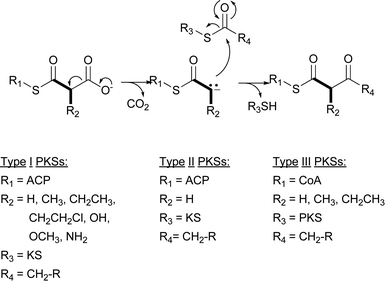 | ||
| Fig. 1 Basic mechanism of decarboxylative Claisen condensations for extender unit incorporation. | ||
Type I PKSs can generally be divided into two groups, modular and iterative. The modular system is the classic bacterial type I PKS best exemplified by the PKS responsible for assembling the 6-deoxyerythronolide B (6-DEB) scaffold of erythromycin A (Fig. 2).6 This PKS assembles seven precursors consisting of one propionyl-CoA starter unit and six (2S)-methylmalonyl-Coenzyme A (CoA) extender units into 6-DEB. The role each of these types of precursors plays in polyketide assembly is readily apparent from the unit names: the starter unit is the initiating precursor for polyketide synthesis, while the extender units elongate the polyketide backbone to completion. A set of catalytic domains, grouped together as a “module,” controls the incorporation of each precursor into the polyketide backbone. For modular type I PKSs, the number of modules is equivalent to the number of precursors incorporated into the polyketide. While the modules that incorporate the starter unit can have variable catalytic domains, the modules that incorporate extender units typically consist of three core domains for polyketide extension and up to three auxiliary domains involved in β-keto processing. The core domains are the ketosynthase (KS), acyltransferase (AT), and acyl carrier protein (ACP). The AT domain is the “gate keeper” of the module and recognizes the specific extender unit to be incorporated into the growing polyketide chain and covalently tethers the malonyl derivative of the extender unit onto the sulfhydryl group of the ACP prosthetic group. This prosthetic group is the 4′-phosphopantetheinyl (4′-Ppant) moiety of CoA. The KS domain catalyzes the decarboxylative Claisen condensation between a neighboring ACP-linked malonate derivative and an ACP-linked acyl thioester to extend the polyketide chain. The remaining optional domains (ketoreductase [KR], dehydratase [DH], and enoylreductase [ER]) alter the oxidation state of the β-keto group formed after the KS-catalyzed condensation. Because its structure is co-linear with the module and domain architecture, the assembly of 6-DEB from one starter unit and six extender units is readily apparent from an analysis of the erythromycin PKS (Fig. 2).
 | ||
| Fig. 2 Erythromycin A-associated modular type I PKS incorporating six (2S)-methylmalonyl-CoA extender units. Labeling above the PKS identifies the loading module (LM) and six modules (M1–M6). The abbreviations for domains are in the text. The carbons in bold represent the extender unit incorporation. | ||
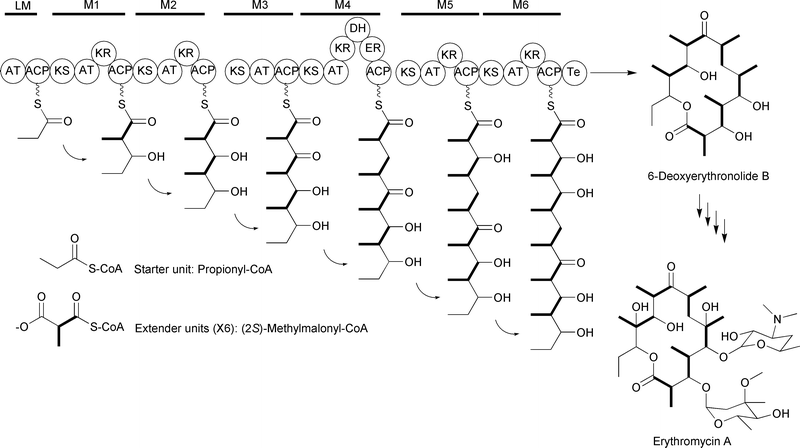
Iterative type I PKSs utilize the same core catalytic domains as modular type I PKSs, but these domains occur on a single polypeptide that is used repetitively to generate the entire polyketide backbone. An example of this type of system is the lovastatin PKS, in which an iteratively acting multidomain polypeptide condenses one starter unit with eight extender units and S-adenosylmethionine (SAM) to generate the dihydromonacolin L intermediate of lovastatin (Fig. 3).7,8 While initially thought to be limited to fungal systems, iterative type I PKSs have now been found in many bacteria as well. Additionally, single modules within modular PKSs have been found to function iteratively to incorporate multiple extender units per polyketide synthesized. Thus, the definition of iterative type I PKSs is expanding, as discussed recently in reviews by Shen,2 Müller,9 and Wilkinson.10
 | ||
| Fig. 3 Lovastatin-associated iterative type I PKS incorporating eight malonyl-CoA extender units. The ER domain of LovB is inactive and replaced by the function of LovC. | ||
Type II PKSs contain similar core catalytic domains seen in type I PKSs, with the exception that there are typically two KS domains, KSα and KSβ. The former is equivalent to the KS seen in type I PKSs, while the latter controls polyketide length, and the enzymatic activities are typically present on individual proteins. No reductive processing of the β-keto groups occurs until after the polyketide is fully synthesized. Classic examples of type II PKS systems are the two “minimal” PKSs involved in generating the anticancer drugs daunorubicin and doxorubicin (Fig. 4).11,12
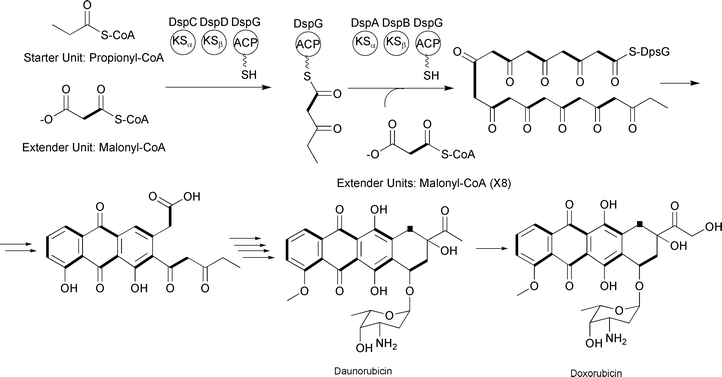 | ||
| Fig. 4 Daunorubicin and doxorubicin-associated type II PKS incorporating nine malonyl-CoA extender units. The KSα enzyme is the KS domain that catalyzes the decarboxylative Claisen condensation of the precursors. The KSβ enzyme controls polyketide length. | ||
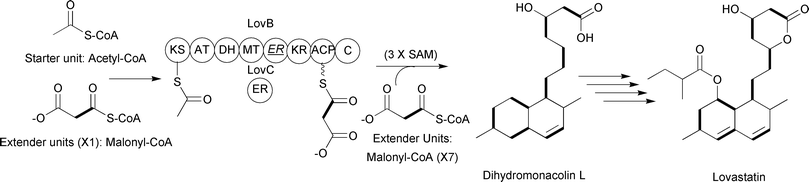
Like type I and type II PKSs, type III PKSs condense a starter unit with a series of extender units to generate a poly-β-keto chain. Type III PKSs differ from the above types in that they typically lack multiple catalytic domains and utilize an ACP-independent mechanism. Instead, a single enzyme harnesses acyl-CoA thioesters and catalyzes acyl group transfers between CoA and an active site cysteine. For example, the type III PKS DpgA generates the hydrated precursor of the dihydroxyphenylacetyl thioester during 3,5-dihydroxyphenylglycine biosynthesis for vancomycin production (Fig. 5).13
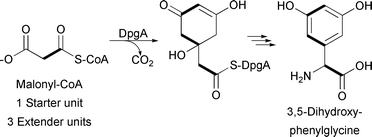 | ||
| Fig. 5 3,5-Dihydroxyphenylglycine-associated type III PKS incorporating three malonyl-CoA extender units. | ||
The enormous structural diversity seen in the polyketide backbones of natural products comes predominantly from the diversity of starter and extender units, variations in the number of extender units incorporated, alterations in the oxidation state of the β-keto groups, and cyclization versus hydrolysis for chain termination. A review by Moore and Hertweck provides an excellent summary of the extensive diversity of starter units utilized by type I and type II PKSs.14 There have also been thorough reviews on the alterations of the oxidation state of the β-keto groups and termination reactions involving cyclization or hydrolysis by thioesterases.3,15 The focus of this review is to provide an update on the biosynthetic pathways that produce the extender units utilized by PKSs, with an emphasis on those extender units found in bacterial systems. This is an appropriate time to review these components of polyketide biosynthesis due to the recent identification of a number of new extender units and the identification of a new metabolic pathway for acetate utilization, the ethylmalonyl-CoA pathway,16 which has implications on the metabolic origins of (2S)-methylmalonyl-CoA and (2S)-ethylmalonyl-CoA extender units. This review is divided into two sections based on the two types of extender units that have been identified. The first section focuses on the biosynthesis of the CoA-linked extender units: malonyl-CoA, (2S)-methylmalonyl-CoA, (2S)-ethylmalonyl-CoA, and chloroethylmalonyl-CoA. The second section focuses on the ACP-linked extender units: (2R)-methoxymalonyl-ACP, (2R)-hydroxymalonyl-ACP, and (2S)-aminomalonyl-ACP (Fig. 6). This second section will also include a brief discussion of the role glyceryl-ACP plays in tetronate ring formation for some polyketides due to the similarities between its biosynthesis and that of (2R)-methoxymalonyl-ACP and (2R)-hydroxymalonyl-ACP.
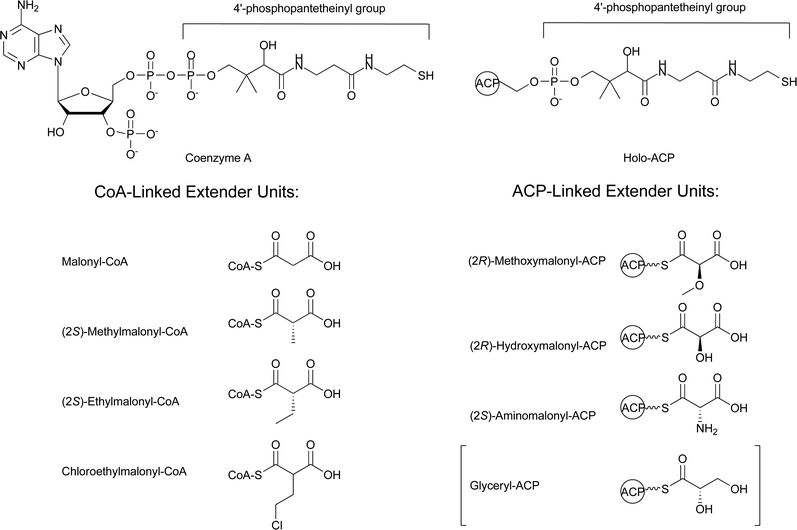 | ||
| Fig. 6 Schematic of CoA, holo-ACP, and the associated extender units. The brackets around glyceryl-ACP denote that it is not a classic extender unit involved in decarboxylative Claisen condensation reactions. | ||
2 Coenzyme A-linked extender units
CoA is essential in both primary and secondary metabolism as an acyl carrier. In polyketide biosynthesis, CoA can play two distinct roles as an acyl carrier. One role is to serve as a substrate for the post-translational modification of ACPs. This modification, catalyzed by phosphopantetheinyltransferases, is the addition of the 4′-Ppant moiety of CoA onto a conserved seryl residue of the ACP to generate a holo-ACP (Fig. 6), the active form of the ACP that functions during polyketide assembly. A second role for CoA in polyketide biosynthesis is as a carrier of carboxylic acid precursors, formally referred to as CoA-linked PKS extender units (Fig. 6). As stated above, the focus of this review is on the biosynthesis of PKS extender units with an emphasis on those observed in bacterial systems. This section focuses on the biosynthesis and occurrence of the four known CoA-linked PKS extender units malonyl-CoA, (2S)-methylmalonyl-CoA, (2S)-ethylmalonyl-CoA, and chloroethylmalonyl-CoA.2.1 Malonyl-CoA
The formation of malonyl-CoA is the first committed step in fatty acid biosynthesis for many organisms; thus, malonyl-CoA is a readily available primary metabolite. Based on the evolutionary relationship of fatty acid and polyketide biosynthesis, it is not surprising that malonyl-CoA is a common extender unit used by all types of PKSs. There are two generally accepted pathways for the formation of malonyl-CoA. The more common pathway involves the carboxylation of acetyl-CoA to generate malonyl-CoA by acetyl-CoA carboxylases (ACCs), while the other involves the direct conversion of malonate to malonyl-CoA by malonyl-CoA synthetase.The ACCs catalyze the carboxylation of acetyl-CoA by a biotin- and ATP-dependent mechanism.17 These enzymes require three components: 1) biotin carboxylase (BC), 2) biotin carboxyl carrier protein (BCCP), and 3) carboxyltransferase (CT). The BC component catalyzes the ATP-dependent activation of bicarbonate and subsequent carboxylation of the N1 atom of the biotin prosthetic group of BCCP (Fig. 7A). Once the carboxyl-BCCP intermediate is formed, the CT activates the methyl group of acetyl-CoA for nucleophilic attack of the activated carboxyl group tethered to BCCP, resulting in the transfer of the carboxyl group onto acetyl-CoA, forming malonyl-CoA (Fig. 7B).
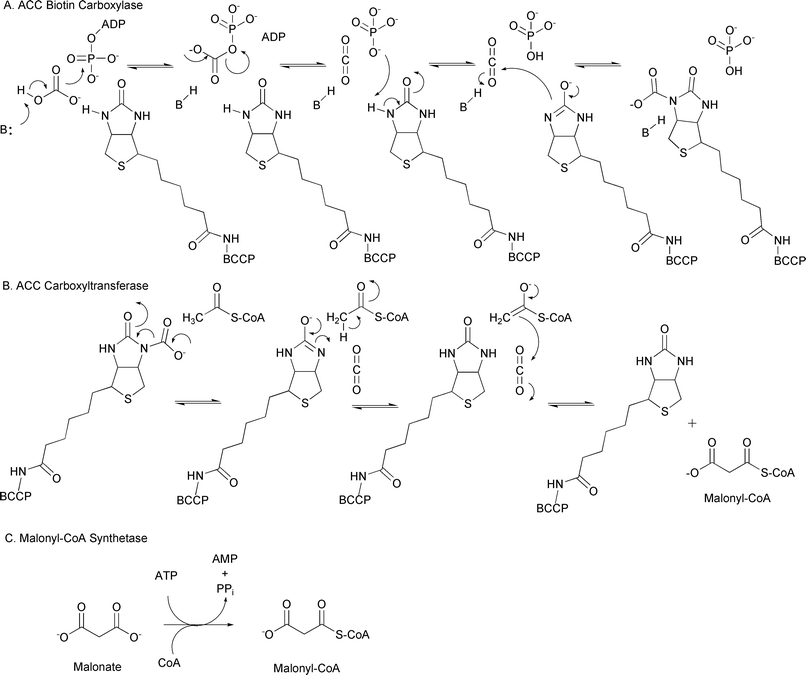 | ||
| Fig. 7 A) Mechanism of carbonate activation and tethering to the biotin cofactor of BCCP; B) Mechanism of the carboxyltransferase reaction generating the extender unit malonyl-CoA; C) The reaction catalyzed by malonyl-CoA synthetase. | ||
While the mechanism of malonyl-CoA biosynthesis by an ACC is conserved throughout the three domains of life, four different forms of the enzyme have been identified. In Escherichia coli, as well as many other bacteria, the three components of ACC are found on four separate proteins.18 The BC and BCCP components are contained on individual polypeptides, AccB and AccC, respectively. The BC forms a homodimer in solution when isolated, while the BCCP is a tetramer.19 The CT component consists of α and β subunits, AccA and AccD in E. coli, which form an α2β2 complex. In contrast to the enzyme architecture seen in E. coli, Streptomyces coelicolor20,21 and other closely related bacteria such as Mycobacterium tuberculosis22 and Saccharopolyspora erythyraea23 have BC and BCCP components contained on a single polypeptide (α subunit, AccA2), and the α and β subunits of the CT component are also a single polypeptide (β subunit, AccB). The AccA2 and AccB polypeptides are likely to form higher oligomeric states with an α6β6 quaternary structure based on the structure of the homolog propionyl-CoA carboxylase.24 Khosla, Gramajo, and colleagues have also identified a third subunit (ε subunit, AccE) in S. coelicolor that forms a complex with the β subunit and enhances the ACC activity.25 Although the precise mechanism for how the ε subunit enhances ACC activity has yet to be elucidated, the prevailing model is that the ε subunit facilitates the formation of the quaternary structure of ACC.20
The importance of ACC to the viability of S. coelicolor is exemplified by the inability to genetically inactivate the gene coding for AccA2 and the finding that an accB mutant strain could only be constructed when long-chain fatty acids were added to the growth medium.20 Importantly, while the addition of long-chain fatty acids to the growth medium enabled the accB mutant to grow, it did not restore the ability of the mutant strain to biosynthesize actinorhodin or the prodiginines. The production of these natural products requires the incorporation of malonyl-CoA extender units by PKS enzymology;26,27 thus, while feeding long-chain fatty acids enables fatty acid biosynthesis for growth, the absence of a source of malonyl-CoA prohibits the biosynthesis of some polyketide natural products. Conversely, the overexpression of accA2, accB, and accE in S. coelicolor results in a significant increase in actinorhodin production.28 These data strongly suggest that ACC plays a direct role in producing the malonyl-CoA extender units needed for the production of actinorhodin, prodiginines, and likely other polyketides in S. coelicolor; however, its most fundamental role is the production of malonyl-CoA extender units for fatty acid biosynthesis.
In contrast to the component organization seen with ACCs from bacteria, a third type of ACC is observed in animals, plants, and yeast.17 These ACCs are single polypeptides divided into three regions. The N-terminal region contains the BC and BCCP components of the ACC, while the C-terminal region contains the α and β portions of the CT component. A central region covering approximately a third of the protein is located between the BC-BCCP and CT components and is only seen in the single polypeptide versions of ACC. The role this region plays in malonyl-CoA formation is currently unknown. Finally, the fourth form of ACC is from the archaeon Metallosphaera sedula and appears to be a fusion of the two types of bacterial ACCs. The BC and BCCP components are contained on separate proteins while the α and β portions of the CT component are fused as a single protein.29
The second mechanism for malonyl-CoA formation is the direct condensation of malonate and CoA by a malonyl-CoA synthetase (Fig. 7C). While ACC appears to be the dominant enzyme for malonyl-CoA formation during fatty acid and polyketide biosynthesis, malonyl-CoA synthetase appears to be involved in the catabolism of malonate in select bacteria.30 Malonate is generated during the degradation of pyrimidines, and it has been proposed that this malonate is converted to malonyl-CoA and subsequently decarboxylated to generate either acetyl-CoA and CO2 or acetate and CO2.31,32 These reactions are likely to be biologically relevant for certain plant-associated bacteria, based on the finding that leguminous plants produce malonate in high levels when they are associated with nitrogen-fixing symbionts, potentially providing a carbon source for the bacteria.33 Consistent with this hypothesis, An and Kim31 identified a gene cluster in Rhizobium trifolii that codes for a dicarboxylate transporter, a malonyl-CoA synthetase, and a malonyl-CoA decarboxylase. These enzymes are proposed to coordinate the transport of malonate into the cell, convert it to malonyl-CoA, and decarboxylate the malonyl-CoA to generate acetyl-CoA and CO2.
While it is not clear whether the malonyl-CoA synthetase makes significant contributions to the malonyl-CoA pool used for polyketide biosynthesis, the malonyl-CoA synthetase is of interest for metabolic engineering purposes. The natural substrate for the enzyme is malonate, but Khosla and colleagues have determined that the enzyme from R. trifolii also recognizes methylmalonate, thereby generating methylmalonyl-CoA.34,35 This provides a simple mechanism for enabling a strain to produce methylmalonyl-CoA from exogenously added methylmalonate. The importance of methylmalonyl-CoA as a PKS extender unit is discussed in the next section.
As stated above, malonyl-CoA is used as an extender unit by all types of PKSs. In all cases, the incorporation of malonyl-CoA into the growing polyketide proceeds via a decarboxylative Claisen condensation between the carboxyl group of a thioester and the enolate ion generated by the decarboxylation of malonyl-CoA. This results in the addition of a two-carbon unit into the polyketide backbone. This can be easily detected by including 13C- or 14C-labeled acetate in the growth medium and analyzing the 13C- or 14C-enrichment pattern. For example, the modular type I PKS that generates the immunosuppressant rapamycin incorporates 14 extender units into the macrocyclic polyketide. Inclusion of [13C2]acetate in the growth medium resulted in the 13C enrichment of C8–C9, C12–C13, C14–C15, C18–C19, C20–C21, C26–C27, and C32–C33, consistent with the incorporation of seven malonyl-CoA extender units into the rapamycin polyketide core (Fig. 8).36 Analysis of the rapamycin biosynthetic gene cluster led to the identification of seven AT domains within the type I PKS showing signature sequences for malonyl-CoA recognition.37 The positions of these malonyl-CoA-dependent AT domains within the PKS are consistent with the location of the acetate units in the rapamycin polyketide backbone identified through the precursor labeling studies.
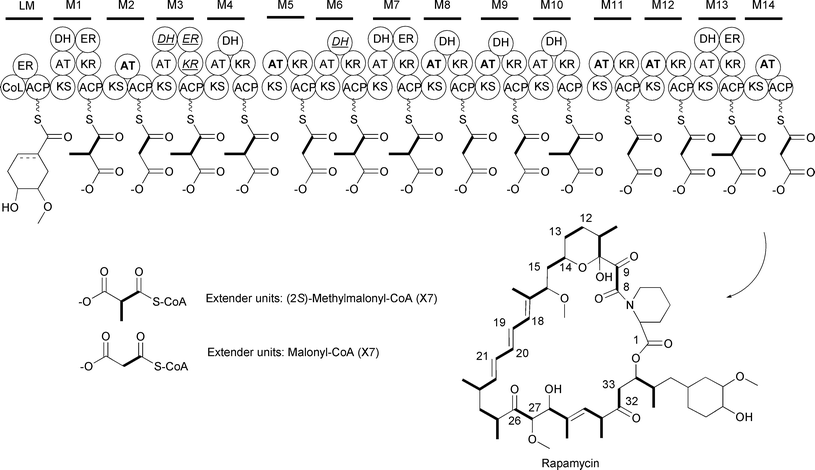 | ||
| Fig. 8 Schematic of the rapamycin modular type I PKS. The AT domains that recognize malonyl-CoA are in bold. The PKS domains that are inactive are italicized and underlined. | ||
Iterative type I PKSs have also harnessed malonyl-CoA extender units for the assembly of polyketides. For example, labeling studies on the esperamicins, produced by the bacterium Actinomadura verrucosospora, revealed that all 14 carbons of the enediyne ring portion of the natural product are derived from seven malonyl-CoA extender units.38 When the biosynthetic gene clusters of the structurally related enediynes calicheamicin39 and C-102740 were identified, the PKS involved in the assembly of the 14-membered ring was found to be an iterative type I PKS contained on a single polypeptide with six distinct domains (Fig. 9). Based on the structural similarities of esperamicin A1 with calicheamicin and C-1027, it has been proposed that the biosynthesis of all of these molecules use malonyl-CoA extender units for the carbons found in the enediyne portion of these natural products.39–41
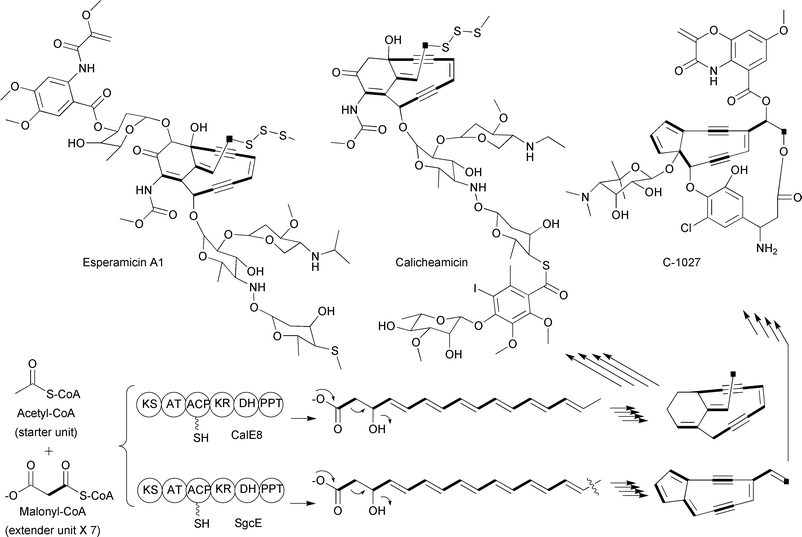 | ||
| Fig. 9 Schematic of the iterative type I PKSs involved in enediyne biosynthesis along with representative enediyne structures. The abbreviations of the enzymatic domains are as described in the text with the addition of PPT, phosphopantetheinyltransferase. | ||
In addition to the modular and iterative type I PKSs, type II and type III PKSs also use malonyl-CoA extender units for assembling the polyketide backbones. The biosynthesis of the anticancer drugs doxorubicin and daunorubicin are assembled by type II PKSs that condense a propionyl-CoA starter unit with nine malonyl-CoA extender units to generate the polyketide backbone of these natural products as discussed previously (Fig. 4). Additionally, the assembly of the nonproteinogenic amino acid 3,5-dihydroxyphenylglycine used in the biosynthesis of the vancomycin-family of antibiotics involves a type III PKS as discussed above (Fig. 5).
2.2 (2S)-Methylmalonyl-CoA
(2S)-Methylmalonyl-CoA is the second most commonly used extender unit for polyketide biosynthesis. As with malonyl-CoA, (2S)-methylmalonyl-CoA incorporation into polyketides is not surprising based on the widespread use of this primary metabolite as an extender unit in fatty acid biosynthesis. For example, Mycobacterium tuberculosis uses (2S)-methylmalonyl-CoA for the biosynthesis of mycolic acids and complex glycolipids for its unusual cell membrane.42 While (2S)-methylmalonyl-CoA is a common primary metabolite, its use as a PKS extender unit is almost exclusively limited to iterative and modular type I PKSs in bacteria. One exemption is the type III PKS from the pine Pinus strobus, PstrCHS2, which has been observed to use a single (2S)-methylmalonyl-CoA extender unit to generate a C-methylated chalcone.43 The incorporation of (2S)-methylmalonyl-CoA extends the polyketide backbone by two carbons, with one carbon bearing a methyl branch, thereby generating a propionyl moiety in the polyketide. While malonyl-CoA incorporated into a polyketide backbone comes predominantly, if not exclusively, from the carboxylation of acetyl-CoA by ACC, (2S)-methylmalonyl-CoA can be generated by a number of mechanisms depending on the growth conditions and the metabolic capability of the producing organism. (2S)-Methylmalonyl-CoA utilized for polyketide biosynthesis can arise from any of the following processes: 1) the carboxylation of propionyl-CoA, 2) the rearrangement and epimerization of succinyl-CoA, 3) the catabolism of valine, or 4) the multistep conversion of acetoacetyl-CoA to (2S)-methylmalonyl-CoA through a crotonyl-CoA-dependent pathway (Fig. 10). Integrated into some of these pathways is the potential incorporation of intermediates from the β-oxidation pathway of fatty acid catabolism. While these are not the only mechanisms by which (2S)-methylmalonyl-CoA can be biosynthesized, the pathways listed above have been linked to the production of polyketide natural products through extensive labeling studies and will be the only pathways discussed here. The metabolic capabilities of the producing organism as well as the carbon source the organism is growing on determine the pathway(s) used for the production of (2S)-methylmalonyl-CoA for polyketide biosynthesis. This variability has made it difficult to fully understand the metabolic origin of the (2S)-methylmalonyl-CoA in some polyketides.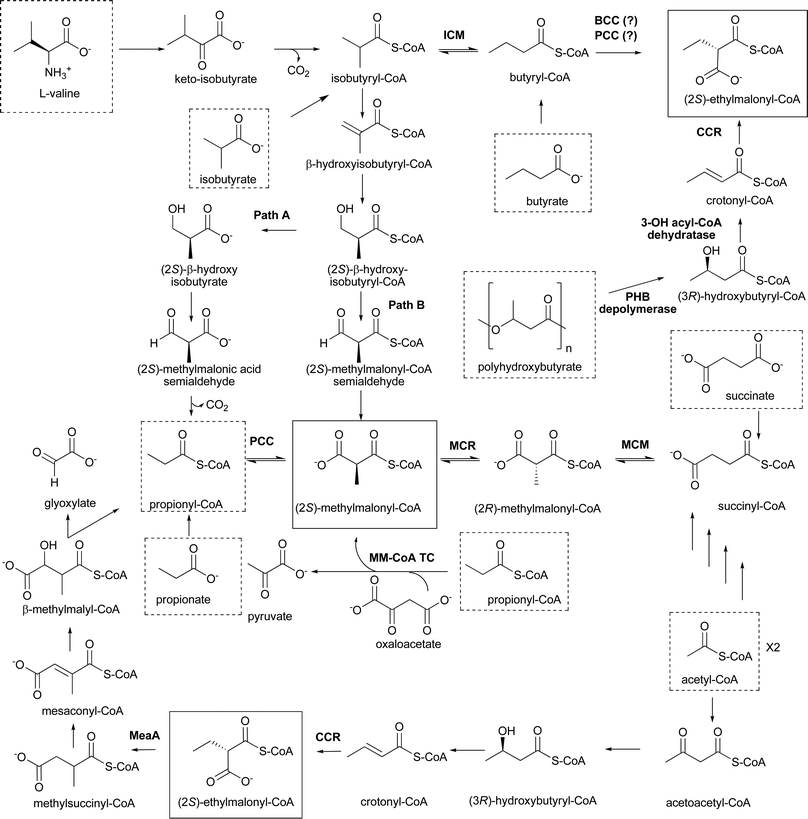 | ||
| Fig. 10 Metabolic pathways leading to the formation of (2S)-methylmalonyl-CoA and (2S)-ethylmalonyl-CoA. These two extender units are in solid boxes; starting metabolites that can lead to these extender units are in dashed boxes. Path A and B are pathways described in the text. Abbreviations: ICM, isobutyryl-CoA mutase; BCC, butyryl-CoA carboxylase; PCC, propionyl-CoA carboxylase; CCR, crotonyl-CoA carboxylase/reductase; PHB, polyhydroxybutyrate; MCR, methylmalonyl-CoA racemase; MCM, methylmalonyl-CoA mutase; MM-CoA TC, methylmalonyl-CoA transcarboxylase. | ||
Propionyl-CoA is frequently the metabolic precursor of (2S)-methylmalonyl-CoA, and it is formed as a product of the β-oxidation of odd- and branched-chain fatty acids; the catabolism of amino acids isoleucine, methionine, and valine; or the breakdown of cholesterol.44,45 Propionyl-CoA can also be formed by the incorporation of propionate from the environment followed by thioesterification with CoA by an acyl-CoA ligase.46 Once formed, propionyl-CoA enters into cellular metabolism through a variety of pathways, one of which is the carboxylation of propionyl-CoA to (2S)-methylmalonyl-CoA. The predominant enzyme that catalyzes this carboxylation is mechanistically similar to the ACC involved in malonyl-CoA formation discussed in the previous section. The propionyl-CoA carboxylase (PCC) is an ATP- and biotin-dependent enzyme complex that catalyzes the activation of carbonate and the decarboxylative condensation between the activated carbonyl moiety and the C2 methyl group of propionyl-CoA. The mechanistic similarity of PCC with ACC is best exemplified by the finding that the α subunit of PCC is the same protein as the α subunit of ACC in S. coelicolor.24 The β subunits of ACC and PCC are distinct and control which acyl-CoA substrate is carboxylated. A second mechanism for propionyl-CoA carboxylation involves methylmalonyl-CoA transcarboxylase.47 This enzyme catalyzes the carboxylation of propionyl-CoA using oxaloacetate as the carboxyl donor to generate (2S)-methylmalonyl-CoA and pyruvate (Fig. 10).
While growth conditions can alter whether propionyl-CoA carboxylation is essential for (2S)-methylmalonyl-CoA formation in the natural producer of a specific polyketide, the PCC enzymes have been exploited in heterologous systems to make this extender unit available in organisms that naturally lack this metabolic capability. For example, the genes encoding the components of the S. coelicolor PCC, along with a propionyl-CoA synthetase, have been introduced into E. coli to enable this bacterium to synthesize (2S)-methylmalonyl-CoA for the heterologous production of erythromycin.48 These same genes have also been introduced into Saccharomyces cerevisiae to enable this model eukaryote to assemble a (2S)-methylmalonyl-CoA-dependent triketide.49 These heterologous systems benefit from the ability to introduce propionate into the culture medium for incorporation into the polyketide, thus circumventing the need to divert important precursors away from primary metabolism for production of the non-native polyketide pathway.
Another precursor to the (2S)-methylmalonyl-CoA extender unit is the primary metabolite succinyl-CoA. One of the most common sources of succinyl-CoA is the tricarboxylic acid cycle (TCA) cycle where succinyl-CoA is an intermediate that can be diverted away for a variety of metabolic purposes including the generation of (2S)-methylmalonyl-CoA. The conversion of succinyl-CoA to (2S)-methylmalonyl-CoA is a two-step process (Fig. 10). First, methylmalonyl-CoA mutase rearranges succinyl-CoA to (2R)-methylmalonyl-CoA in a coenzyme B12-dependent reaction. (2R)-Methylmalonyl-CoA has the incorrect stereochemistry for incorporation into a polyketide, but this problem is corrected by a methylmalonyl-CoA racemase that converts (2R)-methylmalonyl-CoA to (2S)-methylmalonyl-CoA. The importance of succinyl-CoA conversion to (2S)-methylmalonyl-CoA in some organisms is best exemplified by recent work by Leadlay and colleagues which found the gene coding for the methylmalonyl-CoA mutase and genes involved in the biosynthesis of coenzyme B12 are immediately adjacent to the putative elaiophylin biosynthetic gene cluster in Streptomyces sp. DSM4137.50 Elaiophylin biosynthesis requires six (2S)-methylmalonyl-CoA extender units (Fig. 11).
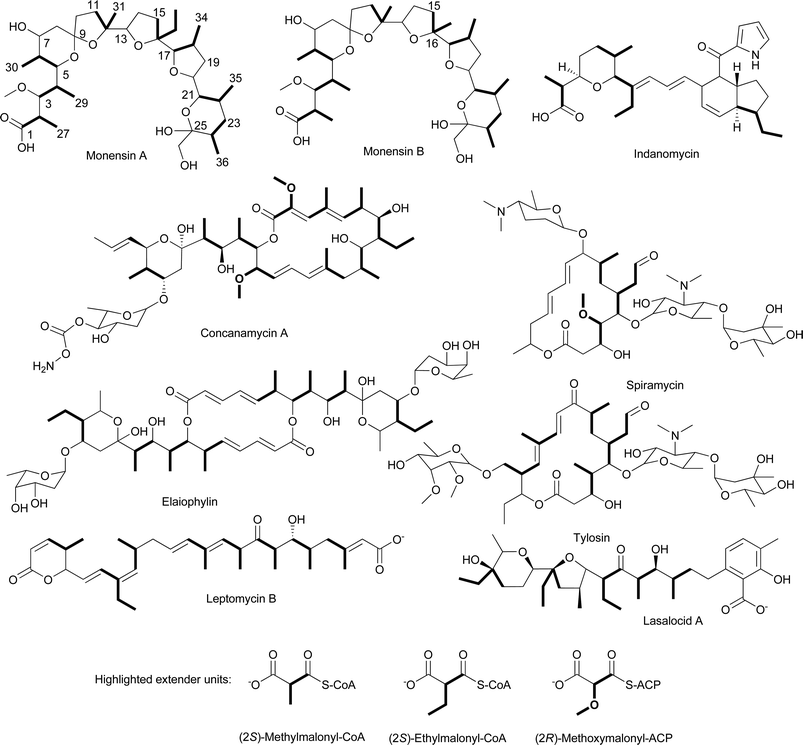 | ||
| Fig. 11 Chemical structures of natural products that incorporate (2S)-methylmalonyl-CoA, (2S)-ethylmalonyl-CoA, or (2R)-methoxymalonyl-ACP extender units. | ||
Determining whether producing organisms use propionyl-CoA or succinyl-CoA as the precursor to (2S)-methylmalonyl-CoA is complicated by the interconnection of these pathways. One pathway for an organism to catabolize propionate is to convert propionate to succinyl-CoA. This pathway involves the carboxylation of propionyl-CoA by PCC and racemization and rearrangement via methylmalonyl-CoA intermediates to succinyl-CoA for entry into the TCA cycle. Thus, in many cases the producing organism can adjust the metabolic source of (2S)-methylmalonyl-CoA according to the growth conditions. For example, extensive labeling studies of the source of (2S)-methylmalonyl-CoA for erythromycin biosynthesis found that either propionate or succinate was incorporated into the macrolide.51,52 Complicating this was the finding that when the gene coding for either the β subunit of PCC53 or the methylmalonyl-CoA mutase54 was independently inactivated, erythromycin was still produced by Saccharopolyspora erythraea. In fact, when a strain of S. erythraea that was lacking a functional methylmalonyl-CoA mutase was grown in a carbohydrate-based medium, erythromycin production was increased by 126% relative to the wild-type strain grown under identical conditions.54 In contrast, when the same mutant was grown in oil-based medium, the amount of erythromycin produced was decreased by 66% relative to the wild-type strain.54 These data highlight the variability in the metabolic origin of (2S)-methylmalonyl-CoA, especially when the growth medium is varied. These data also demonstrate that propionyl-CoA and succinyl-CoA are not the only sources of precursors for (2S)-methylmalonyl-CoA in S. erythraea, an observation that is true in other polyketide-producing organisms as well.
The identification of additional sources of (2S)-methylmalonyl-CoA has come from the extensive studies of polyether and macrolide biosynthesis. One of these sources is the catabolism of the amino acid L-valine.55–61 The initial model for the catabolism of L-valine to (2S)-methylmalonyl-CoA was based on the known pathway in for L-valine degradation to isobutyryl-CoA, which is subsequently converted to propionyl-CoA via a (2S)-β-hydroxyisobutyryl-CoA intermediate. Propionyl-CoA is subsequently carboxylated to (2S)-methylmalonyl-CoA by PCC (Fig. 10, Path A). Labeling studies alone or in combination with mutant analysis have shown this is not the metabolic route utilized for polyether or macrolide assembly in a variety of actinomycetes. For example, labeling studies on monensin A and B, lasalocid A, and tylosin59–61 (Fig. 11) using [1-13C]isobutyrate resulted in 13C enrichment of the C1 of each propionate moiety in the polyketides. If the pathway to (2S)-methylmalonyl-CoA proceeded through propionyl-CoA as initially hypothesized, the label would have been lost as CO2. Furthermore, the addition of [3,3′-13C2]isobutyrate to the culture medium of Streptomyces cinnamonensis resulted in the 13C enrichment of the C27, C29, C30, C31, C34, C35 and C36 carbons of propionyl units of monensin A.60 These data are inconsistent with the initially proposed pathway, which predicted the 13C enrichment of C1, C3, C5, C11, C17, C21, and C23 of the propionyl units (Fig. 11). These data led to the proposal that the isobutyryl-CoA formed from L-valine is converted to (2S)-methylmalonyl-CoA via a methylmalonyl-CoA semialdehyde intermediate (Fig. 10, Path B).62
An additional source of (2S)-methylmalonyl-CoA has been identified in the monensin A and B-producing bacterium S. cinnamonensis. Reynolds and colleagues have performed an extensive analysis of the metabolic origin of the carbons incorporated into monensin A and B when the producing bacterium is grown on distinct carbon sources, providing one of the most thorough analyses of precursor flux into a polyketide.63–69 In these studies, they determined that crotonyl-CoA carboxylase/reductase (CCR) is essential for providing the majority of the (2S)-methylmalonyl-CoA extender units for monensin A and B production when S. cinnamonensis is grown on an oil-based medium.67 CCR is involved in the incorporation of acetyl-CoA generated from the degradation of the fatty acids found in the oil-based medium. In this pathway, two acetyl-CoA units are condensed into acetoacetyl-CoA, followed by the two-step conversion to crotonyl-CoA. Models for how crotonyl-CoA is converted to (2S)-methylmalonyl-CoA were based on the results from the initial in vitro characterization of CCR.70 This work found that CCR catalyzed the reduction of crotonyl-CoA to butyryl-CoA. With butyryl-CoA as an intermediate, the generation of (2S)-methylmalonyl-CoA was proposed to proceed to isobutyryl-CoA, which can enter the same pathway as discussed above for L-valine-derived (2S)-methylmalonyl-CoA (Fig. 10).
Recent data by Alber and colleagues suggest this model needs to be reevaluated.16 Alber and colleagues were investigating an alternative to the glyoxylate cycle for acetyl-CoA assimilation by phototrophic bacteria. They detected a carboxylation reaction (using [14C]carbonate) in extracts of Rhodobacter sphaeroides that were incubated with acetoacetyl-CoA. This carboxylation reaction required the addition of NADPH, and when the carboxylated molecule was purified, it was identified as ethylmalonyl-CoA. Importantly, the carboxylation reaction did not require the addition of ATP, eliminating either ACC or PCC as the possible source of the carboxylation activity. The enzyme catalyzing this activity was determined to be CCR, the enzyme that was previously thought to function solely as a crotonyl-CoA reductase. Alber and colleagues showed that CCR can catalyze the conversion of crotonyl-CoA to butyryl-CoA, but it is more efficient at catalyzing the conversion of crotonyl-CoA to ethylmalonyl-CoA. Thus, CCR is a crotonyl-CoA carboxylase/reductase, not solely a crotonyl-CoA reductase. Once ethylmalonyl-CoA is formed, the pathway would involve the rearrangement to form methylsuccinyl-CoA, which is reduced to mesaconyl-CoA, followed by its hydration to β-methylmalyl-CoA. Finally, a β-methylmalyl-CoA/L-malyl-CoA lyase would generate glyoxylate and propionyl-CoA, with the carboxylation of propionyl-CoA by PCC resulting in the generation of (2S)-methylmalonyl-CoA (Fig. 10). Importantly, Alber and colleagues also analyzed S. coelicolor for CCR activity and showed this organism carries an enzyme with the same enzymatic properties as that seen for the CCR from R. sphaeroides. Therefore, the pathway is likely to function in a similar manner in both R. sphaeroides and S. coelicolor.
In further support of this proposed pathway, Alber and collagues also identified a potential function for MeaA in (2S)-methylmalonyl-CoA formation. Reynolds and colleagues originally identified MeaA as a putative coenzyme-B12-dependent mutase based on its sequence similarity to methylmalonyl-CoA mutase and isobutyryl-CoA mutase.66 The potential role of MeaA in (2S)-methylmalonyl-CoA formation came from the finding that overexpression of meaA in S. cinnamonensis resulted in a significant increase in monensin B production over that of monensin A. Additionally, inactivation of meaA resulted in higher production levels of monensin A compared to monensin B. Monensin A and B differ in whether an ethylmalonyl-CoA or (2S)-methylmalonyl-CoA extender unit is used for C-15 and C-16 (Fig. 11). Clearly, MeaA is not the only source of (2S)-methylmalonyl-CoA, but it does influence the cellular pool of the precursor that is available for polyketide biosynthesis. At the time these studies were done, the identity of the MeaA substrate was unknown. However, it was noted that meaA was located immediately downstream of the gene coding for CCR in S. collinus.71 Alber and colleagues report that MeaA catalyzes the rearrangement of ethylmalonyl-CoA to methylsuccinyl-CoA. This is the metabolic step immediately following the formation of ethylmalonyl-CoA by the CCR (Fig. 10). Thus, CCR and MeaA (ethylmalonyl-CoA mutase) are components of the same pathway that has now has been called the ethylmalonyl-CoA pathway.16 This proposal is consistent with the genetic data discussed above. Inactivating meaA will result in an increase in (2S)-ethylmalonyl-CoA concentrations with the expected increase in monensin A production over monensin B. Overexpression of meaA would likely result in a depletion of (2S)-ethylmalonyl-CoA pools since the increased levels of MeaA will convert the available (2S)-ethylmalonyl-CoA to methylsuccinyl-CoA and then subsequently to (2S)-methylmalonyl-CoA. Thus, the ratio of monensin A and B change accordingly.
While there are a variety of potential sources of (2S)-methylmalonyl-CoA, labeling studies have focused on the use of [13C]propionate to investigate whether the extender unit incorporated into the polyketide backbone is (2S)-methylmalonyl-CoA. The use of [13C]propionate depends on the presence of PCC or alternative carboxylases that are capable of catalyzing propionyl-CoA carboxylation to (2S)-methylmalonyl-CoA. For example, investigations into the precursors used for rapamycin biosynthesis identified seven sites for (2S)-methylmalonyl-CoA incorporation based on the intact incorporation of [13C]propionate into these sites. Consistent with this, the modular type I PKS contains seven modules specific for (2S)-methylmalonyl-CoA (Fig. 8).72 Similarly, labeling studies on erythromycin using [13C]propionate established that one propionyl-CoA starter unit and six (2S)-methylmalonyl-CoA extender units are used by the modular PKS to assemble the macrolide polyketide core (Fig. 2).73
The use of (2S)-methylmalonyl-CoA extender units is also found in iterative type I PKS systems as well. For example, labeling studies on aureothin biosynthesis found that the assembly requires one nitrobenzoate starter unit along with one malonyl-CoA and four (2S)-methylmalonyl-CoA extender units.74 The evidence for the (2S)-methylmalonyl-CoA extender units was based on the observation that the intact [13C]propionate was incorporated into the corresponding sites. Interestingly, sequencing of the aureothin biosynthetic gene cluster suggested the complete type I PKS was “missing” one module.74 Further investigation into the first module of the PKS determined this module works iteratively to incorporate two (2S)-methylmalonyl-CoA extender units. Thus, aureothin is assembled by a combination of an iterative type I PKS module and three modular type I PKS modules to complete the synthesis of the polyketide backbone of aureothin (Fig. 12).
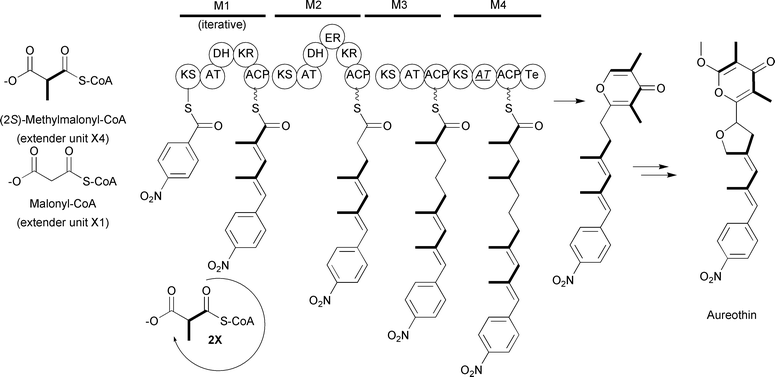 | ||
Fig. 12 Schematic of the type I PKS involved in aureothin biosynthesis. The domain abbreviations are as in the text with the addition of ![[A with combining low line]](https://www.rsc.org/images/entities/i_char_0041_0332.gif) ![[T with combining low line]](https://www.rsc.org/images/entities/i_char_0054_0332.gif) representing an inactive AT domain. representing an inactive AT domain. | ||
2.3 (2S)-Ethylmalonyl-CoA
A number of polyketides synthesized by type I PKSs, including monensin A,75 elaiophylin,76 concanamycin A,77 tylosin,73 leptomycin B,78 spiramycin,79 and indanomycin80 contain a butyryl group within the polyketide core (Fig. 11). More recently, such a moiety was also found to be a component of the germicidin polyketides synthesized by a type III PKS (Fig. 13).81 The butyryl moiety in these, and many other polyketides, can be attributed to the use of the extender unit (2S)-ethylmalonyl-CoA based on labeling studies and biosynthetic gene cluster analyses. The direct source of (2S)-ethylmalonyl-CoA was thought for many years to be solely butyryl-CoA that was carboxylated to form (2S)-ethylmalonyl-CoA. The source of this butyryl-CoA was believed to be through the catabolism of L-valine, the conversion of acetoacetyl-CoA, or the β-oxidation of even-chain fatty acids. As discussed in the prior section, the determination that crotonyl-CoA is directly converted to (2S)-ethylmalonyl-CoA without first going through butyryl-CoA makes a subtle adjustment to one of the proposed pathways leading to (2S)-ethylmalonyl-CoA.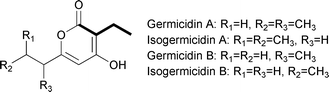 | ||
| Fig. 13 Chemical structures of the germicidins. | ||
One pathway that does proceed through butyryl-CoA has its metabolic origin with L-valine (Fig. 10). As discussed for (2S)-methylmalonyl-CoA formation, L-valine is catabolized to isobutyryl-CoA. Instead of proceeding towards (2S)-methylmalonyl-CoA as discussed previously, isobutyryl-CoA is converted to butyryl-CoA by a coenzyme-B12-dependent isobutyryl-CoA mutase.62 The proposed pathway involves the subsequent carboxylation of butyryl-CoA to (2S)-ethylmalonyl-CoA by an as yet to be identified carboxylase. An additional source of butyryl-CoA is likely to be derived from the β-oxidation of even-chain fatty acids. As with the catabolism of L-valine, a carboxylating enzyme is needed for the conversion of butyryl-CoA to (2S)-ethylmalonyl-CoA. For both the L-valine and β-oxidation pathways, a possible enzyme for this activity is PCC. This hypothesis comes from the finding that Alber and colleagues investigated the source of butyryl-CoA carboxylation in R. sphaeroides and subsequently isolated PCC.16 Thus, the carboxylation of butyryl-CoA was a side activity of PCC. This carboxylation is the most likely reason labeled butyrate added to the culture of a polyketide-producing bacterium results in the intact incorporation of butyrate into the butyryl moieties of the associated polyketides. It is also reasonable to hypothesize that some organisms will code for a butyryl-CoA carboxylase to enhance the levels of (2S)-ethylmalonyl-CoA. One possible example of this occurs in the biosynthesis of the insecticide indanomycin (Fig. 11). Recent work by Kelly and colleagues determined that the indanomycin biosynthetic gene cluster codes for a homolog of the β-subunit of ACC and PCC from Streptomyces species.80 The β-subunit of these enzymes controls substrate specificity; therefore, it is reasonable to speculate that this may code for the CT portion of a butyryl-CoA carboxylase to increase the availability of (2S)-ethylmalonyl-CoA for indanomycin biosynthesis. Further genetic and biochemical studies are needed to test this hypothesis and the alternative hypothesis that this enzyme might be present to increase the levels of (2S)-methylmalonyl-CoA by functioning as part of a PCC.
The two other pathways for (2S)-ethylmalonyl-CoA biosynthesis have their metabolic origin with acetoacetyl-CoA or the β-oxidation of even-chain fatty acids, with both pathways leading to the formation of crotonyl-CoA. This intermediate is converted directly to (2S)-ethylmalonyl-CoA by CCR as discussed for methylmalonyl-CoA (Fig. 10). While the newly identified (2S)-ethylmalonyl-CoA pathway has been proposed as an alternative to the glyoxylate cycle for utilization of acetate as a carbon source, not all polyketide-producing organisms have this pathway. For example, no CCR activity or ccr gene has been detected in the erythromycin-producing bacterium S. erythraea.82 Thus, when the erythromycin pathway was engineered to generate 6-desmethyl-6-ethylerythromycin A, a copy of the gene coding for the CCR from S. collinus had to be introduced into S. erythraea before it would produce the engineered macrolide.82 In contrast, the introduction of the leptomycin gene cluster into S. lividans did not require the addition of a CCR-coding gene for production of the butyryl-containing natural product leptomycin B (Fig. 11), suggesting S. lividans already has the ability to produce (2S)-ethylmalonyl-CoA at sufficient levels for polyketide production.83
Finally, sequencing of the biosynthetic gene cluster for the immunosuppressant FK520 produced by Streptomyces hygroscopicus var. ascomyceticus suggested that this organism uses the storage compound polyhydroxybutyrate as the metabolic origin of (2S)-ethylmalonyl-CoA.84 This hypothesis is based on the FK520 biosynthetic gene cluster coding for homologs of a polyhydroxybutyrate depolymerase and a 3-hydroxyacyl-CoA dehydratase, along with a CCR (Fig. 10). Labeling studies suggest the producing organism also retains the ability to catalyze the carboxylation of butyryl-CoA for (2S)-ethylmalonyl-CoA as well.85S. hygroscopicus var. ascomyceticus is therefore likely to be capable of generating (2S)-ethylmalonyl-CoA from acetoacetyl-CoA, β-oxidation pathway intermediates, or polyhydroxybutyrate (Fig. 10).
While there is more than one pathway to (2S)-ethylmalonyl-CoA, the CCR-dependent pathways may predominate for polyketide production. This proposal is based on the prevalence of genes coding for CCRs within the biosynthetic gene clusters for butyryl-containing polyketides. For example, the elaiophylin,50 tylosin,86 spiramycin,87 concanamycin A,88 and indanomycin80 biosynthetic gene clusters all code for CCR homologs. This may reflect the need to generate excess (2S)-ethylmalonyl-CoA from the catabolism of acetoacetyl-CoA; alternatively, the organism may be siphoning crotonyl-CoA away from the β-oxidation of fatty acids. Consistent with either of these proposals, the concanamycin A, elaiophylin, and indanomycin biosynthetic gene clusters code for not only a CCR, but also a 3-hydroxybutyryl-CoA reductase that potentially reduces acetoacetyl-CoA to 3-hydroxybutyryl-CoA. Most likely, the dehydratase for the conversion of 3-hydroxybutyryl-CoA to crotonyl-CoA comes from primary metabolism resulting in an increase in metabolic flux through crotonyl-CoA to provide sufficient quantities of (2S)-ethylmalonyl-CoA extender units for optimal polyketide production.
The biosynthetic pathways that utilize (2S)-ethylmalonyl-CoA but do not code for a CCR will likely rely on metabolic flux through the ethylmalonyl-CoA pathway for butyrate utilization as proposed by Alber and colleagues.16 Reynolds and colleagues have also shown that there is only one CCR coded by the S. cinnamonensis genome, and the associated gene is adjacent to other genes likely to be associated with the ethylmalonyl-CoA pathway.64 This finding suggests that in organisms such as S. cinnamonensis, flux through the ethylmalonyl-CoA pathway is sufficient for primary metabolism and the formation of polyketides.
2.4 Chloroethylmalonyl-CoA
The extender units discussed up to this point can be involved in both primary metabolism and polyketide biosynthesis. However, chloroethylmalonyl-CoA discussed here and the ACP-linked extender units discussed below are used solely for the production of polyketides. This assertion is based on the failure to find these extender units as part of any specific primary metabolic pathway. Additionally, the biosynthetic enzymes needed for the production of these extender units are coded within the biosynthetic gene cluster for the specific polyketide that contains the extender unit. The utilization of these extender units is much less common than those discussed above; in fact, in some cases they have been found only in a single biosynthetic pathway. The first of these unusual extender units to be discussed is chloroethylmalonyl-CoA, an extender unit that was discovered while investigating the metabolic potential of marine actinomycetes.Moore, Fenical and colleagues at the Scripps Institute for Oceanography have been leaders in the analysis of marine actinomycetes for unique natural products. They have isolated over 2500 members of a new actinomycete genus, Salinospora, which have proven to be prolific producers of natural products with antibiotic and anticancer activities.89 Of particular interest is the production of salinosporamide A that is produced by S. tropica CNB-440 (Fig. 14A).89 This metabolite was originally isolated because of its potent activity against HCT-116 human colon carcinoma, exerting its cytotoxic activity by inactivating the S20 subunit of the proteasome.90,91
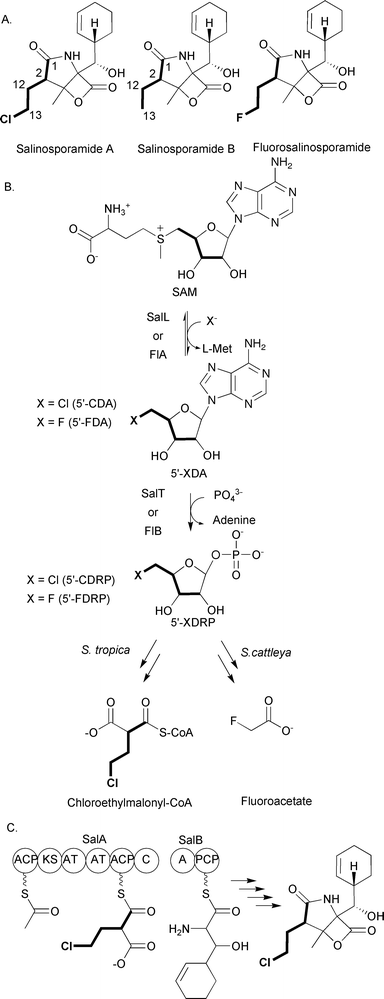 | ||
| Fig. 14 A) Chemical structures of salinosporamide A, salinosporamide B, and fluorosalinosporamide; B) Schematic of chloroethylmalonyl-CoA biosynthesis by S. tropica and fluoroacetate biosynthesis by Streptomyces cattleya; C) Megasynthase involved in chloroethylmalonyl-CoA incorporation and salinosporamide A biosynthesis. | ||
At the core of salinosporamide A is a fused γ-lactam-β-lactone ring structure that is decorated with a cyclohexanyl carbinol at the C4 ring junction, a chloroethyl substituent at C2, and a methyl group at C3. It is the source of the chloroethyl substituent that is of interest for this review. Initial insights into the metabolic origin of this substituent came from comparisons of the biosynthesis of salinosporamide A and a related molecule produced by S. tropica CNB-440, salinosporamide B (Fig. 14A). This related molecule lacks the chlorination on the ethyl substituent on C2, and the initial hypothesis was that salinosporamide B was converted to salinosporamide A based on a recently identified mechanism for halogenations in other natural products.92 The biosynthetic mechanism would involve the formation and incorporation of (2S)-ethylmalonyl-CoA by a polyketide synthase, followed by the chlorination of the ethyl substituent at some later step in the assembly of salinosporamide A. Surprisingly, it has now been determined that the chloroethyl substituent in salinosporamide A and the analogous ethyl substituent in salinosporamide B have different metabolic origins; thus, salinosporamide B is not an intermediate on the pathway to salinosporamide A. Two groups based this conclusion on a series of feeding and labeling studies. While investigating fermentation conditions to enhance the production of salinosporamide B, Lam and colleagues found that the addition of excess butyric acid to the growth medium enhanced production of salinosporamide B by 319% but reduced production of salinosporamide A by 24% relative to wild-type levels.93 Furthermore, addition of [U-13C4]butyrate to the culture medium resulted in an enrichment of 13C in the four-carbon unit C1, C2, C12, and C13 of salinosporamide B, but not in the corresponding carbons of salinosporamide A.
At approximately the same time, Beer and Moore performed an extensive set of feeding experiments with stable isotopes to investigate the metabolic origin of each component of salinosporamide A and salinosporamide B.94 They also observed that the addition of 13C-labeled butyrate to the culture medium did not result in the enrichment of 13C into the chloroethyl substituent of salinosporamide A, but enrichment was observed in the corresponding carbons of salinosporamide B. Furthermore, feeding of [1,2-13C2]acetate resulted in the 13C-enrichment of the carbons in the ethyl substituent of salinosporamide B, but not the chloroethyl substituent of salinosporamide A. Thus, formation of the chloroethyl substituent does not involve the formation and incorporation of ethylmalonyl-CoA followed by chlorination. Beer and Moore went on to investigate the enrichment of the C1, C2, C12, and C13 carbons in salinosporamide A using [U-13C6]glucose and [1,3-13C2]glycerol. From these studies it was hypothesized that the metabolic source of these carbons in salinosporamide A is a sugar. The intact incorporation of the four carbons from a single [U-13C6]glucose to form the C1, C2, C12, and C13 carbons suggests the involvement of the pentose phosphate pathway in generating the sugar precusor. Based on these data, Beer and Moore proposed the existence of a previously unknown precursor pathway that formed the PKS extender unit chloroethylmalonyl-CoA. This precursor would be incorporated into salinosporamide A, while ethylmalonyl-CoA would replace the chlorethylmalonyl-CoA to generate salinosporamide B.
While these labeling studies were essential in providing insights into how the cell assembles salinosporamide A and salinosporamide B, it was not until the biosynthetic gene cluster associated with the production of these natural products was uncovered that a clear picture appeared for how the chloroethyl substituent is likely to be assembled. Moore and colleagues sequenced the genome of S. tropica CNB-440 and discovered a 41-kb gene cluster coding for 29 ORFs they proposed to be involved in the production of salinosporamide A and salinosporamide B.95,96 One of the key findings was that while the cluster does not code for a homolog of the recently characterized oxidative chlorinating enzymes,92 it does code for a homolog of the fluorinase FlA involved in fluoroacetate production. FlA is an unusual enzyme that catalyzes the conversion of SAM to 5′-fluoro-5′-deoxyadenosine (5′-FDA) (Fig. 14B).97 Processing of 5′-FDA eventually leads to the formation of fluoroacetate production.98 Moore and colleagues hypothesized that the initial steps in chloroethylmalonyl-CoA formation follow a similar route based on a FlA homolog being coded by the salinosporamide A and salinosporamide B biosynthetic gene cluster. Indeed, Moore, Noel, and colleagues showed that SalL, the FlA homolog, catalyzes the conversion of SAM to 5′-chloro-5′-deoxyadenosine (5′-CDA), analogous to the formation of 5′-FDA (Fig. 14B).96 In support of these in vitro data, a S. tropica CNB-440 strain that is deleted for salL continues to produce salinosporamide B but fails to produce salinosporamide A.99
The conversion of 5′-CDA to chloroethylmalonyl-CoA remains to be fully elucidated, but it is clear the next step will likely follow that seen for fluoroacetate formation. Briefly, FlB has been shown to catalyze the conversion of 5′-FDA to 5-fluoro-5-deoxy-D-ribose-1-phosphate (5′-FDRP) (Fig. 14B).98 A homolog of FlB, SalT, is coded by the biosynthetic gene cluster for salinosporamide A and salinosporamide B. Thus, it was proposed that SalT enzyme will catalyze the conversion of 5′-CDA to 5-chloro-5-deoxy-D-ribose-1-phosphate (5′-FDRP).99 Based on this proposal, Moore and colleagues recently used mutasynthesis to show that feeding 5′-FDA or 5′-FDRP to a salL mutant results in the formation of fluorosalinosporamide (Fig. 14A), consistent with the mutasynthesis of an unnatural extender unit fluoroethylmalonyl-CoA. These data are consistent with the current hypothesis that chloroethylmalonyl-CoA is derived from SAM and proceeds through both 5′-CDA and 5′-CDRP. On-going studies by the Moore group continue to investigate this fascinating metabolic pathway.
The incorporation of chloroethylmalonyl-CoA into salinosporamide A is controlled by a mixed PKS/NRPS megasynthase with unusual domain architecture. Briefly, salA codes for a six-domain megasynthase that is proposed to incorporate acetyl-CoA and chloroethylmalonyl-CoA (or ethylmalonyl-CoA and fluoroethylmalonyl-CoA) (Fig. 14C). Once the diketide is formed, it is likely condensed with the cyclohexenyl alanine moiety tethered to SalB, a didomain nonribosomal peptide synthetase. What makes the SalA domain architecture unusual is that it does not follow the standard KS-AT-ACP module organization commonly seen with modular type I PKSs. Instead, the megasynthase initiates with an ACP domain that is likely acetylated by the AT domain located at the center of the protein adjacent to the AT domain that incorporates chloroethylmalonyl-CoA or ethylmalonyl-CoA. A single KS domain likely condenses the acetyl and chloroethylmalonyl groups and leaves the diketide tethered to the second AT domain. The C-terminal condensation domain of SalA could then transfer the diketide to SalB for further processing to salinosporamide A or B. At this time, the salinosporamide system is the only PKS system known to use chloroethylmalonyl-CoA as an extender unit.
3 Acyl carrier protein-linked extender units
The next class of PKS extender units differs from the CoA-linked extender units discussed above in that they are covalently tethered to the 4′-Ppant groups of holo-ACPs (Fig. 6). Also, unlike malonyl-CoA, (2S)-methylmalonyl-CoA, and (2S)-ethylmalonyl-CoA, these precursors are unique to polyketide biosynthesis, with the genes coding for their biosynthesis found within or flanking the associated biosynthetic gene clusters. Although they are not nearly as common as malonyl-CoA or (2S)-methylmalonyl-CoA extender units, they are becoming more prevalent as more natural product biosynthetic pathways are elucidated. Currently, they are all found in pathways involving modular type I PKSs, although future natural product discoveries may reveal their presence in other types of PKSs as well.(2R)-Methoxymalonyl-ACP, (2R)-hydroxymalonyl-ACP, and (2S)-aminomalonyl-ACP elongate a polyketide chain with methoxyacetyl, glycolyl, or glycyl units, respectively, with the substituents at the α-carbons conferring functionalities and hydrogen-bonding potential not available through the use of the CoA-linked extender units.84,100 While it has not been described as a PKS extender unit, another ACP-linked PKS precursor worthy of discussion is glyceryl-ACP, which incorporates a glycerol-derived three-carbon unit into a tetronate ring of polyketides known as acyltetronic acids.101 The occurrence and biosynthesis of these four precursors is discussed below.
3.1 (2R)-Methoxymalonyl-ACP
The first ACP-linked PKS extender unit to be reported was (2R)-methoxymalonyl-ACP, which results in the incorporation of a methoxyacetyl group into a polyketide.84 First proposed as an extender unit in the biosynthesis of FK520 by Streptomyces hygroscopicus, (2R)-methoxymalonyl-ACP has since been proposed as a precursor in a number of other polyketides including ansamitocin P-3,102 bafilomycin A1,,103 concanamycin A,88 geldanamycin,104 herbimycin,105 leucomycin,106 midecamycin,107 niddamycin,108 oxazolomycin,109 soraphen A,110 and tautomycin,111 all of which have α-carbons bearing methoxyl groups (Figs. 11, 15). Feeding experiments performed with the FK520-producer and 13C-labeled precursors demonstrated that this unusual moiety is derived not from acetate, methoxyacetate, or methoxymalonate but rather from an intermediate of glycolysis.112,113 Feeding experiments performed with a number of other polyketides containing this unusual moiety are consistent, suggesting the involvement of a “glycolate” extender unit.103,106,114–119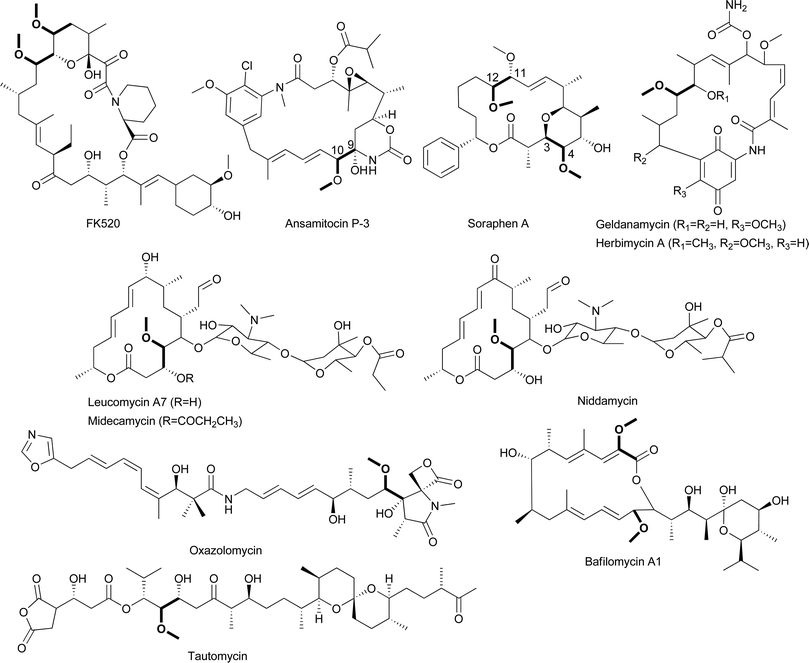 | ||
| Fig. 15 Chemical structures of natural products that incorporate (2R)-methoxymalonyl-ACP. | ||
To investigate the possibility that this “glycolate” extender unit is derived from a CoA-linked thioester such as hydroxymalonyl-CoA or methoxymalonyl-CoA, Floss and colleagues fed the ansamitocin P-3 producer 13C-labeled N-acetyl-cysteamine (SNAC) thioesters,120 which have been shown to be cell-permeable and effective as in vivo and in vitro mimics of CoA thioesters.35,121,122 After feeding the producer [1-13C](2R)-hydroxymalonyl-SNAC and [1-13C](2R)-methoxymalonyl-SNAC thioesters, they observed no 13C enrichment. Similar experiments performed by Grond and Schuhmann103 with the bafilomycin A1 and concanamycin A producers also resulted in no 13C enrichment. These data suggested that hydroxymalonyl-CoA and methoxymalonyl-CoA are unlikely precursors of the methoxyacetyl moiety.
Significant insight into the identity of this precursor was gained from the sequencing and analysis of the biosynthetic gene cluster for FK520. Reeves and colleagues proposed that a subset of five genes encoding the enzymes FkbG, FkbH, FkbI, FkbJ, and FkbK were involved in the formation of the extender unit (2R)-methoxymalonyl-ACP.84 All of the enzymes except FkbH were homologous to proteins of known function: FkbG was homologous to plant caffeoyl-CoA O-methyltransferases; FkbI, acyl-CoA dehydrogenases; FkbJ, ACPs; and FkbK, 3-hydroxyacyl-CoA dehydrogenases. In their proposal, FkbH covalently tethers a glycolytic pathway intermediate to the 4′-Ppant group of holo-FkbJ, forming glyceryl-FkbJ. Next, FkbK oxidizes glyceryl-FkbJ to an aldehyde intermediate, and FkbI further oxidizes this intermediate to (2R)-hydroxymalonyl-FkbJ. As a final step, FkbG catalyzes the O-methylation of (2R)-hydroxymalonyl-FkbJ to form (2R)-methoxymalonyl-FkbJ (Fig. 16A).
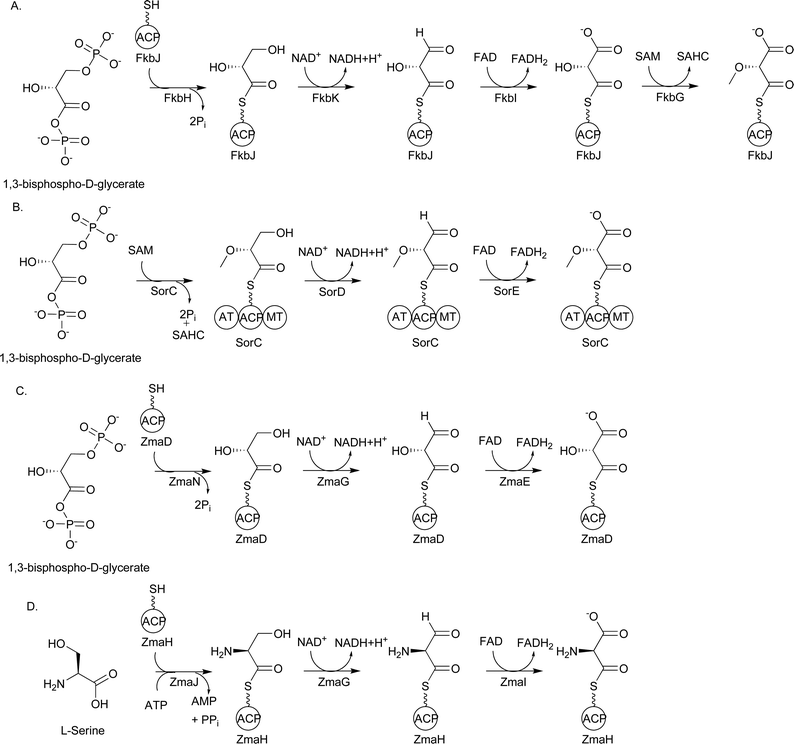 | ||
| Fig. 16 A) Proposed biosynthetic pathway for FkbH-dependent (2R)-methoxymalonyl-ACP formation; B) Proposed biosynthetic pathway for (2R)-methoxymalonyl-ACP formation during soraphen A biosynthesis; C) Biosynthetic pathway for (2R)-hydroxymalonyl-ACP formation during zwittermicin A biosynthesis; D) Biosynthetic pathway for (2S)-aminomalonyl-ACP formation during zwittermicin A biosynthesis. SAHC = S-adenosylhomocysteine. | ||
In support of this proposal, genes coding homologs to these five enzymes have been found in the biosynthetic pathways for a number of other methoxyacetyl-containing natural products, including ansamitocin P-3, concanamycin A1, geldanamycin, herbimycin, oxazolomycin, spiramycin, and tautomycin.87,88,104,105,109,111 Interestingly, the biosynthetic gene cluster for soraphen A codes for homologs to FkbK and FkbI but lacks a gene coding an FkbH homolog; instead, sorC codes a three-domain protein containing an AT, ACP, and methyltransferase (MT) domain (Fig. 16B).110 The AT domain is predicted to covalently tether the glycolytic substrate to the ACP domain of SorC, forming glyceryl-SorC. The predicted dehydrogenases SorD and SorE may oxidize this intermediate to (2R)-hydroxymalonyl-SorC, which is then O-methylated by the MT domain of SorC. The presence of the MT domain on the same polypeptide as the ACP led Floss and colleagues to propose that glyceryl-SorC is first O-methylated to form 2-O-methyl-glyceryl-SorC, which is then oxidized to (2R)-methoxymalonyl-SorC. By extension, Floss et al. argue that O-methylation likely precedes the oxidation steps during ansamitocin biosynthesis as well.114 Further work is necessary to establish the order of the oxidation/methylation events, which may differ from pathway to pathway.
Work by Floss and colleagues has provided genetic evidence supporting the proposal that this subset of five genes is involved in (2R)-methoxymalonyl-ACP formation. In the ansamitocin biosynthetic pathway, the genes asm13–17 code homologs to FkbK, J, I, H, and G, respectively.123 A mutation in asm15 eliminated ansamitocin production, though small amounts of the derivative 10-desmethoxy-ansamitocin were produced as a result of the misincorporation of a malonyl-CoA extender unit in the absence of the “glycolate” extender unit.120 Supplementing the asm15 mutant with (2R)-hydroxymalonyl-SNAC or (2R)-methoxymalonyl-SNAC thioesters did not restore ansamitocin production, consistent with previous experiments in which labeled SNAC thioesters were not incorporated by the wild-type producer. The lack of incorporation of SNAC thioesters is also consistent with the notion of an ACP-linked versus CoA-linked precursor. Further evidence for the involvement of an ACP came from an asm14 insertional mutant that was abolished for the production of ansamitocin and any ansamitocin-related compounds; this suggests that the ACP is an essential component for not only the incorporation of the methoxymalonate unit but also the misincorporation of malonate units. These mutagenesis experiments show that at least asm14 and asm15 are necessary for the incorporation of a methoxymalonyl unit. In another set of experiments, a cassette containing asm13–17 was introduced into a vector co-expressing an eryAI, II, III construct coding the 6-DEB PKS with its (2S)-methylmalonyl-CoA-specific AT6 domain replaced with the putative hydroxymalonyl-/methoxymalonyl-ACP-specific AT8 domain from fkbA.113,124 Heterologous expression of the modified 6-DEB vector alone resulted in the production of 6-DEB and 2-desmethyl-6-DEB. Co-expression of the asm13–17 cassette abolished production of these compounds but resulted in the production of the novel compound 2-desmethyl-2-methoxy-6-DEB, in which a (2R)-methoxymalonyl extender unit was incorporated.113,124 To probe whether O-methylation occurs before or after incorporation of the “glycolate” extender unit, a significant portion of the MT-coding gene asm17 was deleted. Co-expression of the modified 6-DEB vector with a cassette containing the mutated asm17 gene resulted in the production of 6-DEB and 2-desmethyl-6-DEB only. The lack of 2-desmethyl-2-hydroxy-6-DEB suggested that O-methylation occurs prior to incorporation by the PKS and that the unmethylated precursor is not competent for incorporation by the PKS. Taken together, these data demonstrate that a cassette containing asm13–17 is sufficient for forming the “glycolate” extender unit, which is likely (2R)-methoxymalonyl-ACP.
Additional genetic evidence that this subset of five genes is involved in (2R)-methoxymalonyl-ACP biosynthesis came from Katz et al. and their work on the heterologous production of midecamycin in Streptomyces fradiae.107 Midecamycin, an antibiotic made by Streptomyces mycarofaciens, contains a methoxyacetyl moiety proposed to be derived from (2R)-methoxymalonyl-ACP. When the midecamycin PKS genes are expressed in the S. fradiae host, no production of midecamycin or midecamycin analogs occurs; however, the integration of fkbGHIJK into the S. fradiae chromosome confers the host strain the ability to produce a midecamycin analog bearing a methoxyacetyl unit in its polyketide backbone.
These genetic data have been supported by biochemical investigations involving enzymes from the FK520 biosynthetic pathway. In 2003, Tsai and colleagues solved the crystal structure for FkbI and proposed that its structure supports its role as an acyl-ACP versus acyl-CoA dehydrogenase.125 This proposal was based upon docking simulations of FkbI with a homology model of FkbJ as well as sequence comparisons of FkbI and its homologs to acyl-CoA dehydrogenases. In this work, FkbG, FkbH, FkbJ, and FkbK, were also heterologously overproduced and purified, although no biochemical characterization of these four enzymes was reported at the time. In more recent work, FkbGHIJK were heterologously co-overproduced and purified in an E. coli strain containing sfp, which codes for a PPTase that post-translationally modifies apo-ACPs to their active holo forms.126 To determine if E. coli expressing the fkbGHIJK cassette could produce (2R)-methoxymalonyl-FkbJ in vivo, FkbJ was further purified from the other Fkb enzymes, digested with trypsin, and analyzed by LC/MS. In addition to peaks with masses corresponding to FkbJ + 2H+ and FkbJ + H+ + Na+, a peak with a mass corresponding to (2R)-methoxymalonyl-FkbJ + 2H+ was observed, and the presence of this peak was dependent upon the co-expression of fkbGHIK. Based on a comparison of the peak area ratios, it was estimated that 20% of FkbJ was converted to (2R)-methoxymalonyl-FkbJ. Similar experiments were performed in which FkbGHIK were heterologously co-overproduced with GdmJ, the FkbJ homolog from geldanamycin biosynthesis. LC/MS analysis of trypsin-digested purified GdmJ revealed that FkbGHIK were capable of forming (2R)-methoxymalonyl-GdmJ in vivo. Interestingly, about 60% of GdmJ was converted to (2R)-methoxymalonyl-GdmJ by the Fkb enzymes.
While these data provided biochemical evidence of (2R)-methoxymalonyl-ACP formation in vivo, they did not identify the glycolytic substrate of FkbH. Insights into the identity of this substrate were gained from a sequence analysis of FkbH and its homologs.127 The N-terminal domain contains the motif DXDX(T/V) as well as other conserved residues found in phosphatases belonging to the haloacid dehalogenase (HAD) superfamily of hydrolases; the C-terminal domain contains the conserved SCR motif, consistent with acyltransferases. The AT-like domain was proposed to transfer the glycolytic substrate, an activated carboxylic acid, to the 4′-Ppant group of a holo-ACP. This substrate is likely already activated at its carboxyl group, due to the absence of any obvious carboxyl-activating domains or NTP-binding motifs in the C-terminal domain of FkbH or in any other enzymes associated with the methoxymalonyl-ACP subcluster. Using this metabolic logic, the common glycolytic intermediate 1,3-bisphospho-D-glycerate (1,3-bPG) was proposed to be the substrate for FkbH, with its N-terminal domain responsible for removing the phosphate from the phosphoester group of C3.127
In their investigations with OzmB, the FkbH homolog from the oxazolomycin biosynthetic pathway, Shen and colleagues provided biochemical evidence supporting the proposal that 1,3-bPG is in fact the glycolytic precursor to (2R)-methoxymalonyl-ACP.109 Using HPLC-based assays and MS, they showed that a glyceryl moiety becomes tethered to OzmB in the presence of 1,3-bPG. MS/MS analysis revealed that the addition of this moiety is localized to the CRVV amino acid sequence of OzmB and that glycerate is presumably covalently bound as a thioester to cysteine. To determine if OzmB could transfer this glyceryl moiety to an ACP, they incubated glyceryl-OzmB with holo-TtmD (an ACP from tautomycin bioynthesis). HPLC and MS analysis confirmed the formation of glyceryl-TtmD, suggesting that OzmB was competent for acylating a non-cognate ACP. To investigate the timing of the phosphatase activity of OzmB, they mutated the active site aspartate of the N-terminal HAD domain to a valine and performed the same loading experiments with 1,3-bPG. MS analysis of the resulting product yielded a mass that was consistent with 3-phosphoglyceryl-OzmB. These experiments clearly demonstrate that 1,3-bPG is loaded onto OzmB, forming 3-phosphoglyceryl-OzmB, which is then dephosphorylated to form glyceryl-OzmB, an important first step in forming (2R)-methoxymalonyl-ACP.
The biochemical evidence that 1,3-bPG is the metabolic precursor of the (2R)-methoxymalonyl group is supported most recently by a series of in vivo labeling experiments by Floss, Müller, and colleagues on ansamitocin and soraphen A biosynthesis.114 The ansamitocin P-3 producer, Actinosynnema pretiosum, was fed D-[1,2-13C2]glucose, and the ansamitocin was purified and analyzed by 13C-NMR. This analysis determined that there was no C–C coupling between C9–C10 carbons of the methoxyacetyl group, in contrast to the coupling seen for the carbons originating from malonyl-CoA (Fig. 15). These data are consistent with the hypothesis that the methoxyacetyl group is derived from a triose phosphate or phosphoglycerate precursor originating from the glycolytic pathway.
In further investigations, feeding A. pretiosum [1,2-13C2]glycerate failed to result in any 13C-enrichment, but the addition of [1,2-13C2]glycerate to the culture of Sorangium cellulosum resulted in the expected enrichment at C3–C4 and C11–C12 of the methoxyacetyl groups of soraphen A (Fig. 15). In S. celluosum D-[1,2-13C2]glycerate was preferentially incorporated over L-[1,2-13C2]glycerate in soraphen A, consistent with the involvement of the glycolytic pathway in generating the metabolic precursor to (2R)-methoxymalonyl-ACP since only phospho-D-glycerates are intermediates in the glycolytic pathway. The failure to detect any 13C-enrichment in any carbons of ansamitocin likely reflects the inability of A. pretiosum to phosphorylate exogenously added glycerate for incorporation into the glycolytic pathway.
Finally, using R-[1,2-13C2]glycerol and S-[1,2-13C2]glycerol, Floss, Müller and colleagues investigated whether one stereoisomer was preferentially incorporated over the other in ansamitocin P-3 and soraphen A. The incorporation of S-[1,2-13C2]glycerol over R-[1,2-13C2]glycerol would provide strong evidence that a glycolytic intermediate such as 1,3-bPG is the metabolic origin of (2R)-methoxymalonyl-ACP. As predicted, 13C-enrichment in both A. pretiosum and S. cellulosum required S-[1,2-13C2]glycerol, supporting the role of the glycolytic pathway. These in vivo data, combined with the in vitro biochemical data support the conclusion that 1,3-bPG is the precursor for (2R)-methoxymalonyl-ACP. Importantly, due to the stereospecificity of the glycolytic pathway, the C2 carbon of the (2R)-methoxymalonyl-ACP has the opposite stereochemistry as seen in (2S)-methylmalonyl-CoA and (2S)-ethylmalonyl-CoA. Thus, the incorporation of (2R)-methoxymalonyl-ACP by the associated PKSs requires an AT domain capable of recognizing a protein-linked extender unit that is structurally and stereochemically distinct from the CoA-linked extender units.
3.2 (2R)-Hydroxymalonyl-ACP
Another ACP-linked extender unit, (2R)-hydroxymalonyl-ACP (Figs. 16C, 17), is a proposed precursor in the biosynthesis of (2R)-methoxymalonyl-ACP and is thus formed in a similar way. Reeves and colleagues, in their paper describing their proposals for (2R)-methoxymalonyl-ACP formation in the FK520 biosynthetic pathway, first proposed that (2R)-hydroxymalonyl-ACP could be an additional extender unit in the biosynthesis of aflastatin A, an inhibitor of aflatoxin production.84,128 Previously, labeling studies with 13C-labeled precursors and the streptomycete aflastatin A producer were performed to gain insight into the biosynthetic origin of this unusual polyketide, which has five hydroxyl groups in its long alkyl chain in positions that are atypical for polyketides.128 Feeding the producer 13C-labeled acetate, propionate, glucose, and glycolate, Ono and colleagues determined that while most of the alkyl chain is derived from acetate and propionate, there are five two-carbon units derived from glycolate. The finding that these unusual moieties are derived from a glycolytic intermediate rather than acetate or propionate is consistent with the proposal for a novel enzymatic mechanism involving (2R)-hydroxymalonyl-ACP as an extender unit. It is expected that the sequencing and analysis of the biosynthetic gene clusters for aflastatin A and blasticidin A,129 a structurally related antibiotic and inhibitor of aflatoxin production, will reveal genes encoding homologs of four of the five enzymes involved in (2R)-methoxymalonyl-ACP formation. The presence of hydroxyl versus methoxyl groups at the α-carbons suggests that a gene encoding a methyltransferase will be absent.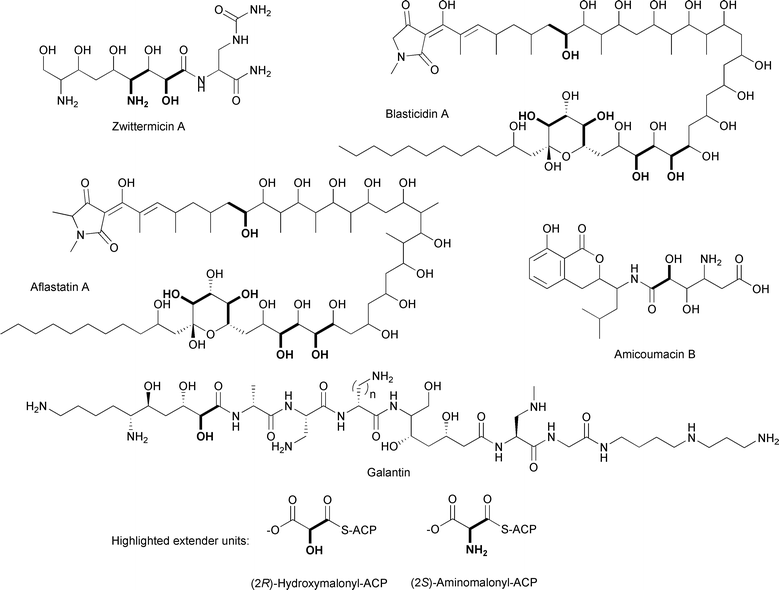 | ||
| Fig. 17 Chemical structures of natural products incorporating (2R)-hydroxymalonyl-ACP or (2S)-aminomalonyl-ACP extender units. | ||
An example of a biosynthetic gene cluster containing genes encoding four of the five enzymes involved in (2R)-methoxymalonyl-ACP formation is the cluster for zwittermicin A (Fig. 17), an antibiotic first identified in Bacillus cereus.130 The presence of the genes zmaN, zmaD, zmaG, and zmaE, encoding homologs of FkbH, FkbJ, FkbK, and FkbI, respectively, as well as an unusual glycolyl moiety in the chemical structure led to the proposal that (2R)-hydroxymalonyl-ACP is used as a PKS extender unit in the biosynthesis of zwittermicin A (Fig. 16).100,131 While no labeling studies with 13C-labeled glycolytic precursors have been performed, the pathway for (2R)-hydroxymalonyl-ACP formation has been reconstituted in vitro using heterologously purified zwittermicin A enzymes.100 Prior to these experiments, which were concurrent with work in the Shen lab on (2R)-methoxymalonyl-ACP formation, the substrate for the FkbH homolog was unknown, although 1,3-bPG was the most likely candidate.84,127 This proposed substrate was shown to be the correct one, and HPLC-based assays and mass spectrometry were used to show that ZmaN dephosphorylates 1,3-bPG while covalently tethering it to the 4′-Ppant group of holo-ZmaD, forming glyceryl-ZmaD. The NAD+-dependent enzyme ZmaG converts glyceryl-ZmaD to 2-hydroxy-3-oxopropionyl-ACP, the substrate for the FAD-dependent dehydrogenase ZmaE, which then converts this intermediate to (2R)-hydroxymalonyl-ZmaD. All of the components of the pathway are required for (2R)-hydroxymalonyl-ZmaD formation. Importantly, MS/MS analysis showed that the tethering and modification of the glycolytic substrate was localized to the amino acid sequence GYVNS of ZmaD, with the serine residue being the site of 4′-Ppant modification. Additional 4′-Ppant elimination experiments confirmed the identity of the covalently-linked extender unit.
Other natural products that have glycolyl units include the linear aminopolyol antibiotic galantin and the pseudopeptide herbicide amicoumacin B, both produced by Bacillus species (Fig. 17).132,133 While these glycolyl units may be derived from a post-assembly tailoring enzyme such as a P450 monooxygenase, labeling studies and/or sequencing efforts may reveal the involvement of (2R)-hydroxymalonyl-ACP in forming these unusual moieties.
3.3 (2S)-Aminomalonyl-ACP
A second ACP-linked PKS extender unit also found in zwittermicin A biosynthesis is (2S)-aminomalonyl-ACP (Figs. 16D, 17). The unusual ethanolamine moiety in zwittermicin A suggested that an atypical extender unit might be involved.131 An examination of the biosynthetic gene cluster revealed the presence of genes encoding additional homologs to (2R)-methoxymalonyl-ACP enzymes, including a carrier protein (ZmaH) and a dehydrogenase (ZmaI). The presence of a gene encoding an adenylation domain with a specificity code for L-serine suggested that instead of a glycolytic precursor, L-serine is covalently tethered to the ACP to form seryl-ZmaH. The FkbK and FkbI homologs ZmaG and ZmaI were proposed to catalyze the two-step oxidation of seryl-ZmaH to (2S)-aminomalonyl-ZmaH in reactions analogous to the oxidation of glyceryl-ZmaD to (2R)-hydroxymalonyl-ZmaD (Fig. 16C).Although no labeling studies with 13C-labeled serine have been performed, (2S)-aminomalonyl-ACP has been reconstituted in vitro using heterologously purified biosynthetic enzymes.100 ATP/PPi-exchange assays demonstrated the specificity of the A domain ZmaJ for L-serine, and HPLC-based assays coupled with MS provided evidence for the formation of (2S)-aminomalonyl-ACP. ZmaJ was shown to activate and covalently tether L-serine to the ACP ZmaH, forming seryl-ZmaH. The dehydrogenases ZmaG and ZmaI were also shown to oxidize seryl-ZmaH in two-steps to form (2S)-aminomalonyl-ACP, an unstable product that spontaneously decarboxylates under the reaction conditions used to form glycyl-ZmaH. Nevertheless, the formation of glycyl-ZmaH is indicative of the formation of (2S)-aminomalonyl-ZmaH. Furthermore, all the components of the pathway (L-serine, ATP, ZmaH, ZmaJ, ZmaG, and ZmaI) were required for (2S)-aminomalonyl-ZmaH formation. Importantly, MS/MS analysis showed that the tethering and modification of the seryl substrate was localized to the sequence GLVNS, with the final serine being the site of 4′-Ppant addition. Analysis of the substituent tethered to the 4′-Ppant prosthetic group through 4′-Ppant elimination experiments confirmed the mass changes.
Interestingly, the zwittermicin A cluster only encodes one FkbK homolog (ZmaG), and this enzyme functions in the biosynthetic pathways for both (2R)-hydroxymalonyl-ACP and (2S)-aminomalonyl-ACP to catalyze the first oxidation step of the glyceryl- or seryl-tethered moieties. When the specificities of the FkbI homologs were tested in each pathway, it was discovered that ZmaE functions in the (2R)-hydroxymalonyl-ACP pathway, whereas ZmaI could function in both the (2R)-hydroxmalonyl-ACP and (2S)-aminomalonyl-ACP pathways. Substrate selectivity was more stringent with ZmaN and ZmaJ, however, as each could only acylate/aminoacylate its cognate ACP.
To date, zwittermicin A represents the only known example in which aminomalonyl-ACP is found as a PKS extender unit. It also represents the only known instance in which NRPS components (an A domain and a carrier protein) are used to form a type I PKS extender unit. We propose that (2S)-aminomalonyl-ACP may also act as an NRPS extender unit in the biosynthesis of other natural products. One such example is the cyclic heptapeptide antibiotic GE23077, which contains an aminomalonic moiety with its amino group in peptidic linkage with an iso-serine residue and one of its carboxyl groups in peptidic linkage with an acylated L-2,3-diaminopropionate residue (Fig. 18).134 Although the biosynthetic gene cluster has not yet been reported, it is possible that a (2S)-aminomalonyl-ACP NRPS extender unit is involved.
 | ||
| Fig. 18 Chemical structure of GE23077A. | ||
3.4 Glyceryl-ACP
Glyceryl-ACP is not technically considered to be a extender unit because it does not elongate the main linear chain of polyketides but rather adds three-carbon units that branch from the main chain. However, because its biosynthesis is so similar to that of (2R)-methoxymalonyl-ACP and (2R)-hydroxymalonyl-ACP and it occurs as an intermediate in the biosynthesis of these extender units, a discussion of glyceryl-ACP is included in this review. Glyceryl-ACP is found as a precursor in the biosynthesis of certain acyltetronic acids, polyketides containing an unusual five-membered tetronate (4-hydroxy-[5H]furan-2-one) ring, though not all acyltetronic acids are believed to be derived from this precursor.127 Labeling studies performed with the antitumor antibiotic chlorothricin show that while most of the structure is derived from acetate and propionate, the three-carbon unit consisting of C22, C23, and C24 is derived from glycerol (Fig. 19).135 Feeding experiments involving other acyltetronic acids such as tetrocarcin A and tetronasin (Fig. 19) are also consistent with a glycolytic precursor.136–138 | ||
| Fig. 19 Chemical structures of natural products that incorporate glyceryl-ACP precursors and a proposed scheme for glyceryl-ACP incorporation into tetronomycin. | ||
The sequencing and analysis of the chlorothricin biosynthetic gene cluster in 2006 revealed the presence of genes coding for homologs of FkbH and FkbJ, suggesting the involvement of an ACP-linked precursor.139 In addition, genes encoding a putative pyruvate dehydrogenase/dehydratase and a putative hydrolase/AT were also found and were proposed to modify and incorporate the ACP-linked precursor into the tetronate group. Parallel work with the kijanimicin and tetronomycin biosynthetic gene clusters also revealed subclusters encoding FkbH homologs and other enzymes proposed to form and incorporate the tetronate groups of these polyketides (Fig. 19).140,141 The proposals for tetronate-ring formation in these three natural products are similar in that they all involve the formation of glyceryl-ACP as a precursor: the FkbH homolog covalently tethers the glycolytic intermediate (1,3-bPG) to an ACP, forming glyceryl-ACP.
In 2007, Leadlay and colleagues reconstituted the pathway for glyceryl-ACP formation in vitro using heterologously purified enzymes from the tetronomycin biosynthetic pathway.101 HPLC-based assays and MS were used to show that the FkbH homolog Tmn16 covalently tethers the substrate 1,3-bPG to the holo-ACP Tmn7a and dephosphorylates it, forming glyceryl-Tmn7a. The cross-reactivity of the Tmn proteins with enzymes from the chlorothricin and concanamycin pathways was also examined. Interestingly, Tmn16 was capable of acylating the ACP homolog ChlD2 from the chlorothricin pathway but not Con2 from the concanamycin pathway. Con2 could be acylated by its cognate FkbH homolog Con4, but Con4 was not capable of acylating either Tmn7a or ChlD2. These results indicate that substrate flexibility may be more likely to occur for enzymes of pathways that are more related (i.e. tetronate polyketides) versus those that are less related (i.e. methoxyacetyl-containing polyketides). Another intriguing observation is that the FkbH homologs Tmn16 and ChlD1 have highly conserved, long N-terminal extensions not present in the other FkbH homologs involved in (2R)-methoxymalonyl-ACP formation (i.e. Con4), and this N-terminal extension was found to be critical for proper function of Tmn7a. It is possible that this feature may contribute to substrate selectivity.
For chlorothricin, tetronomycin, and kijanimicin (Fig. 19) biosynthesis, the pathway for glyceryl-ACP formation is predicted to occur similarly; the proposals for how this precursor is incorporated into the tetronate ring, however, vary from pathway to pathway. For example, in chlorothricin biosynthesis, it has been proposed that glyceryl-ACP is converted to enoylpyruvoyl-ACP, the substrate for a putative hydrolase/AT that incorporates it into the chlorothricin aglycone.139 Meanwhile, in kijanimicin biosynthesis, an AT domain of a putative bifunctional enzyme is predicted to convert glyceryl-ACP to glyceryl-CoA prior to incorporation into the ACP-linked kijanimicin precursor.140 The proposal for tetronomycin formation is that glyceryl-ACP is directly incorporated without prior modification.141 Further investigations are necessary to determine the mechanism of incorporation of this unusual ACP-linked precursor.
4 Other potential extender units
Clearly, the number of PKS extender units has expanded recently from the initial model of the use of only malonyl-CoA and (2S)-methylmalonyl-CoA. While the identification of ACP-linked extender units has expanded the number of extender units considerably, the real expansion of the available extender units may come from those biosynthesized by a mechanism similar to that seen for crotonyl-CoA conversion to (2S)-ethylmalonyl-CoA. The finding by Alber and colleagues that CCR catalyzes not only the reduction of crotonyl-CoA but also its carboxylation raises the possibility that previously unknown extender units could be generated by an analogous reaction on any 2,3-desaturated acyl-CoA thioester. Since these types of intermediates are found during each cycle of the β-oxidation pathway for fatty acid degradation, any of these intermediates could be converted to a PKS extender unit. For example, the structure of FK506 contains an allyl side chain at position C21 (Fig. 20). It has been proposed that a five-carbon PKS extender unit, potentially propylmalonyl-CoA, would function to introduce the carbon unit into the growing polyketide chain.142 Such an extender unit could be formed by the reductive carboxylation of 2-pentenyl-CoA, an intermediate of the β-oxidation of odd-chain fatty acids. | ||
| Fig. 20 Chemical structures of FK506 and polyoxypeptin A which potentially incorporate propylmalonyl-CoA and 2-(2-methyl-butyl)-malonyl-CoA extender units, respectively. | ||
There are possibilities not only with straight-chain fatty acid degradation but also with branched-chain fatty acids. For example, polyoxypeptin A contains a methylbutyl group associated with its alkyl side chain (Fig. 20). This side chain may potentially come from a 2-(2-methyl-butyl)-malonyl-CoA extender unit. The metabolic origin of this extender unit would be the β-oxidation of anteiso-fatty acids that are commonly found in Gram-positive organisms. Feeding the producing Streptomyces strain with L-[13C]isoleucine resulted in the 13C-enrichment of the C7, C8, C8-Me, C9, and C10 carbons of polyoxypeptin A.143 This result is consistent with anteiso-fatty acids as the source of this portion of the polyketide since isoleucine is the source of the branched portion of these fatty acids. The presence of a CCR homolog that recognizes the 4-methyl-2-hexanoyl-CoA would provide a source of this extender unit.
5 Acknowledgements
Research on the biosynthesis of PKS extender units is generously funded by the National Institutes of Health (AI065850) and by an Alfred Toepfer Faculty Fellow to M.G.T. We thank Elizabeth Felnagle for helpful comments. We thank Dr. Wendy L. Kelly (Georgia Institute of Technology) for sharing unpublished data.6 References
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC
.
- B. Shen, Curr. Opin. Chem. Biol., 2003, 7, 285–295 CrossRef CAS
- S. Smith and S. C. Tsai, Nat. Prod. Rep., 2007, 24, 1041–1072 RSC
- M. B. Austin and J. P. Noel, Nat. Prod. Rep., 2003, 20, 79–110 RSC
- C. Hertweck, A. Luzhetskyy, Y. Rebets and A. Bechthold, Nat. Prod. Rep., 2007, 24, 162–190 RSC
- B. J. Rawlings, Nat. Prod. Rep., 2001, 18, 190–227 RSC
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS
- S. M. Ma and Y. Tang, FEBS J., 2007, 274, 2854–2864 CrossRef CAS
- R. Müller, Chem. Biol., 2004, 11, 4–6 CAS
- S. J. Moss, C. J. Martin and B. Wilkinson, Nat. Prod. Rep., 2004, 21, 575–593 RSC
- W. R. Strohl, M. L. Dickens, V. B. Rajgarhia, A. J. Wooand N. D. Priestley, in Biotechnology of Antibiotics, ed. Strohl, Marcel Dekker, New York, 2nd edn., 1997, vol. 82, pp. 577–657 Search PubMed
- S. L. Otten, K. J. Stutzman-Engwall and C. R. Hutchinson, J. Bacteriol., 1990, 172, 3427–3434 CAS
- H. Chen, C. C. Tseng, B. K. Hubbard and C. T. Walsh, Proc. Natl. Acad. Sci. USA, 2001, 98, 14901–14906 CrossRef CAS
- B. S. Moore and C. Hertweck, Nat. Prod. Rep., 2002, 19, 70–99 RSC
- J. Grünewald and M. A. Marahiel, Microbiol. Mol. Biol. Rev., 2006, 70, 121–146 CrossRef
- T. J. Erb, I. A. Berg, V. Brecht, M. Müller, G. Fuchs and B. E. Alber, Proc. Natl. Acad. Sci. USA, 2007, 104, 10631–10636 CrossRef CAS
- L. Tong, Cell. Mol. Life Sci., 2005, 62, 1784–1803 CrossRef CAS
- J. E. Cronan Jr. and G. L. Waldrop, Prog. Lipid Res., 2002, 41, 407–435 CrossRef
- E. Choi-Rhee and J. E. Cronan, J. Biol. Chem., 2003, 278, 30806–30812 CrossRef CAS
- E. Rodríguez, C. Banchio, L. Diacovich, M. J. Bibb and H. Gramajo, Appl. Environ. Microbiol., 2001, 67, 4166–4176 CrossRef CAS
- G. Gago, D. Kurth, L. Diacovich, S. C. Tsai and H. Gramajo, J. Bacteriol., 2006, 188, 477–486 CrossRef CAS
- D. L. Rainwater and P. E. Kolattukudy, J. Bacteriol., 1982, 151, 905–911 CAS
- A. R. Hunaiti and P. E. Kolattukudy, Arch. Biochem. Biophys., 1982, 216, 362–371 CAS
- L. Diacovich, D. L. Mitchell, H. Pham, G. Gago, M. M. Melgar, C. Khosla, H. Gramajo and S. C. Tsai, Biochemistry, 2004, 43, 14027–14036 CrossRef CAS
- L. Diacovich, S. Peirú, D. Kurth, E. Rodríguez, F. Podestá, C. Khosla and H. Gramajo, J. Biol. Chem., 2002, 277, 31228–31236 CrossRef CAS
- F. Malpartida and D. A. Hopwood, Mol. Gen. Genet., 1986, 205, 66–73 CrossRef CAS
- A. M. Cerdeño, M. J. Bibb and G. L. Challis, Chem. Biol., 2001, 8, 817–829 CrossRef CAS
- Y. G. Ryu, M. J. Butler, K. F. Chater and K. J. Lee, Appl. Environ. Microbiol., 2006, 72, 7132–7139 CrossRef CAS
- M. Hügler, R. S. Krieger, M. Jahn and G. Fuchs, Eur. J. Biochem., 2003, 270, 736–744 CrossRef CAS
- Y. S. Kim, J. Biochem. Mol. Biol., 2002, 35, 443–451 Search PubMed
- J. H. An and Y. S. Kim, Eur. J. Biochem., 1998, 257, 395–402 CrossRef CAS
- J. H. An, G. Y. Lee, J. W. Jung, W. Lee and Y. S. Kim, Biochem. J., 1999, 344 Pt 1, 159–166 CrossRef
- Y. S. Kim and S. K. Bang, J. Biol. Chem., 1985, 260, 5098–5104 CAS
- F. Lombó, B. Pfeifer, T. Leaf, S. Ou, Y. S. Kim, D. E. Cane, P. Licari and C. Khosla, Biotechnol. Prog., 2001, 17, 612–617 CrossRef CAS
- N. L. Pohl, R. S. Gokhale, D. E. Cane and C. Khosla, J. Am. Chem. Soc., 1998, 120, 11206–11207 CrossRef CAS
- N. L. Paiva, A. L. Demain and M. F. Roberts, J. Nat. Prod., 1991, 54, 167–177 CrossRef CAS
- T. Schwecke, J. F. Aparicio, I. Molnár, A. König, L. E. Khaw, S. F. Haydock, M. Oliynyk, P. Caffrey, J. Cortés, J. B. Lester and et al., Proc. Natl. Acad. Sci USA, 1995, 92, 7839–7843 CAS
- K. S. Lam, J. A. Veitch, J. Golik, B. Krishman, S. E. Klohr, K. J. Volk, S. Forenza and T. W. Doyle, J. Am. Chem. Soc., 1993, 115, 12340–12345 CrossRef CAS
- J. Ahlert, E. Shepard, N. Lomovskaya, E. Zazopoulos, A. Staffa, B. O. Bachmann, K. Huang, L. Fonstein, A. Czisny, R. E. Whitwam, C. M. Farnet and J. S. Thorson, Science, 2002, 297, 1173–1176 CrossRef CAS
- W. Liu, S. D. Christenson, S. Standage and B. Shen, Science, 2002, 297, 1170–1173 CrossRef CAS
- W. Liu, J. Ahlert, Q. Gao, E. Wendt-Pienkowski, B. Shen and J. S. Thorson, Proc. Natl. Acad. Sci. USA, 2003, 100, 11959–11963 CrossRef CAS
- R. S. Gokhale, P. Saxena, T. Chopra and D. Mohanty, Nat. Prod. Rep., 2007, 24, 267–277 RSC
- J. Schröder, S. Raiber, T. Berger, A. Schmidt, J. Schmidt, A. M. Soares-Sello, E. Bardshiri, D. Strack, T. J. Simpson, M. Veit and G. Schröder, Biochemistry, 1998, 37, 8417–8425 CrossRef CAS
- G. Gottschalk, Bacterial Metabolism, 2nd edn, Springer-Verlag, New York, 1985 Search PubMed
- R. Van der Geize, K. Yam, T. Heuser, M. H. Wilbrink, H. Hara, M. C. Anderton, E. Sim, L. Dijkhuizen, J. E. Davies, W. W. Mohn and L. D. Eltis, Proc. Natl. Acad. Sci. USA, 2007, 104, 1947–1952 CrossRef CAS
- A. R. Horswill and J. C. Escalante-Semerena, J. Bacteriol., 1999, 181, 5615–5623 CAS
- D. B. Northrop, J. Biol. Chem., 1969, 244, 5808–5819 CAS
- B. A. Pfeifer, S. J. Admiraal, H. Gramajo, D. E. Cane and C. Khosla, Science, 2001, 291, 1790–1792 CrossRef CAS
- S. C. Mutka, S. M. Bondi, J. R. Carney, N. A. Da Silva and J. T. Kealey, FEMS Yeast Res., 2006, 6, 40–47 CrossRef CAS
- S. F. Haydock, T. Mironenko, H. I. Ghoorahoo and P. F. Leadlay, J. Biotechnol., 2004, 113, 55–68 CrossRef CAS
- S. M. Friedman, T. Kaneda and J. W. Corcoran, J. Biol. Chem., 1964, 239, 2386–2391 CAS
- A. A. Hunaiti and P. E. Kolattukudy, Antimicrob. Agents Chemother., 1984, 25, 173–178 CAS
- S. Donadio, M. J. Staver and L. Katz, Mol. Microbiol., 1996, 19, 977–984 CrossRef CAS
- A. R. Reeves, I. A. Brikun, W. H. Cernota, B. I. Leach, M. C. Gonzalez and J. M. Weber, J. Ind. Microbiol. Biotechnol., 2006, 33, 600–609 CrossRef CAS
- S. Pospísil, E. Královcová, K. Stajner, J. Tax, V. Krumphanzl and Z. Vaněk, Folia Microbiol., 1982, 27, 275–277 Search PubMed
- S. Ōmura, K. Tsuzuki, Y. Tanaka, H. Sakakibara, M. Aizawa and G. Lukacs, J. Antibiot., 1983, 36, 614–616 CAS
- A. Leiser, A. Birch and J. A. Robinson, Gene, 1996, 177, 217–222 CrossRef CAS
- L. Tang, Y. X. Zhang and C. R. Hutchinson, J. Bacteriol., 1994, 176, 6107–6119 CAS
- S. Pospísil, P. Sedmera, M. Havránek, V. Krumphanzl and Z. Vaněk, J. Antibiot., 1983, 36, 617–619 CAS
- K. A. Reynolds, D. O'Hagan, D. Gani and J. A. Robinson, J. Chem. Soc. Perkin Trans. I, 1988, 3195–3207 RSC
- M. M. Sherman, S. Yue and C. R. Hutchinson, J. Antibiot., 1986, 39, 1135–1143 CAS
- J. W. Vrijbloed, K. Zerbe-Burkhardt, A. Ratnatilleke, A. Grubelnik-Leiser and J. A. Robinson, J. Bacteriol., 1999, 181, 5600–5605 CAS
- D. O'Hagan, S. V. Rogers, G. R. Duffin and K. A. Reynolds, J. Antibiot., 1995, 48, 1280–1287 CAS
- H. Liu and K. A. Reynolds, J. Bacteriol., 1999, 181, 6806–6813 CAS
- H. Liu and K. A. Reynolds, Metab. Eng., 2001, 3, 40–48 CrossRef CAS
- W. Zhang and K. A. Reynolds, J. Bacteriol., 2001, 183, 2071–2080 CrossRef CAS
- C. Li, G. Florova, K. Akopiants and K. A. Reynolds, Microbiology, 2004, 150, 3463–3472 CrossRef CAS
- C. Li, K. Akopiants and K. A. Reynolds, J. Ind. Microbiol. Biotechnol., 2006, 33, 75–83 CrossRef CAS
- K. Akopiants, G. Florova, C. Li and K. A. Reynolds, J. Ind. Microbiol. Biotechnol., 2006, 33, 141–150 CrossRef CAS
- K. K. Wallace, Z. Y. Bao, H. Dai, R. Digate, G. Schuler, M. K. Speedie and K. A. Reynolds, Eur. J. Biochem., 1995, 233, 954–962 CAS
- L. Han and K. A. Reynolds, J. Bacteriol., 1997, 179, 5157–5164 CAS
- P. Caffrey, D. J. Bevitt, J. Staunton and P. F. Leadlay, FEBS Lett., 1992, 304, 225–228 CrossRef CAS
- S. Ōmura, H. Takeshima, A. Nakagawa, J. Miyazawa, F. Piriou and G. Lukacs, Biochemistry, 1977, 16, 2860–2866 CrossRef CAS
- J. He and C. Hertweck, Chem. Biol., 2003, 10, 1225–1232 CrossRef CAS
- L. E. Day, J. W. Chamberlin, E. Z. Gordee, S. Chen, M. Gorman, R. L. Hamill, T. Ness, R. E. Weeks and R. Stroshane, Antimicrob. Agents Chemother., 1973, 4, 410–414 CAS
- M. Gerlitz, P. Hammann, R. Thiericke and J. Rohr, J. Org. Chem., 1992, 57, 4030–4033 CrossRef CAS
- K. U. Bindseil and A. Zeeck, Liebigs Ann. Chem., 1994, 305–312 CrossRef CAS
- T. Hamamoto, T. Uozumi and T. Beppu, J. Antibiot., 1985, 38, 533–535 CAS
- A. Inoue, T. Deguchi, M. Yoshida and K. Shirahata, J. Antibiot., 1983, 36, 442–444 CAS
- W. L. Kelly. Personal communication Search PubMed.
- L. Song, F. Barona-Gomez, C. Corre, L. Xiang, D. W. Udwary, M. B. Austin, J. P. Noel, B. S. Moore and G. L. Challis, J. Am. Chem. Soc., 2006, 128, 14754–14755 CrossRef CAS
- D. L. Stassi, S. J. Kakavas, K. A. Reynolds, G. Gunawardana, S. Swanson, D. Zeidner, M. Jackson, H. Liu, A. Buko and L. Katz, Proc. Natl. Acad. Sci. USA, 1998, 95, 7305–7309 CrossRef CAS
- Z. Hu, R. Reid and H. Gramajo, J. Antibiot., 2005, 58, 625–633 CrossRef CAS
- K. Wu, L. Chung, W. P. Revill, L. Katz and C. D. Reeves, Gene, 2000, 251, 81–90 CrossRef CAS
- K. M. Byrne, A. Shafiee, J. B. Nielsen, B. Arison, R. L. Monaghan and L. Kaplan, Dev. Ind. Microbiol., 1993, 32, 29–45 Search PubMed
- A. R. Gandecha, S. L. Large and E. Cundliffe, Gene, 1997, 184, 197–203 CrossRef CAS
- F. Karray, E. Darbon, N. Oestreicher, H. Dominguez, K. Tuphile, J. Gagnat, M. H. Blondelet-Rouault, C. Gerbaud and J. L. Pernodet, Microbiology, 2007, 153, 4111–4122 CrossRef CAS
- S. F. Haydock, A. N. Appleyard, T. Mironenko, J. Lester, N. Scott and P. F. Leadlay, Microbiology, 2005, 151, 3161–3169 CrossRef CAS
- R. H. Feling, G. O. Buchanan, T. J. Mincer, C. A. Kauffman, P. R. Jensen and W. Fenical, Angew. Chem. Int. Ed. Engl., 2003, 42, 355–357 CrossRef CAS
- D. Chauhan, L. Catley, G. Li, K. Podar, T. Hideshima, M. Velankar, C. Mitsiades, N. Mitsiades, H. Yasui, A. Letai, H. Ovaa, C. Berkers, B. Nicholson, T. H. Chao, S. T. Neuteboom, P. Richardson, M. A. Palladino and K. C. Anderson, Cancer Cell, 2005, 8, 407–419 CrossRef CAS
- M. Groll, R. Huber and B. C. Potts, J. Am. Chem. Soc., 2006, 128, 5136–5141 CrossRef CAS
- F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Chem. Rev., 2006, 106, 3364–3378 CrossRef
- G. Tsueng, K. A. McArthur, B. C. Potts and K. S. Lam, Appl. Microbiol. Biotechnol., 2007, 75, 999–1005 CrossRef CAS
- L. L. Beer and B. S. Moore, Org. Lett., 2007, 9, 845–848 CrossRef CAS
- D. W. Udwary, L. Zeigler, R. N. Asolkar, V. Singan, A. Lapidus, W. Fenical, P. R. Jensen and B. S. Moore, Proc. Natl. Acad. Sci. USA, 2007, 104, 10376–10381 CrossRef CAS
- A. S. Eustáquio, F. Pojer, J. P. Noel and B. S. Moore, Nat. Chem. Biol., 2008, 4, 69–74 CrossRef CAS
- C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O'Hagan and J. H. Naismith, Nature, 2004, 427, 561–565 CrossRef CAS
- F. Huang, S. F. Haydock, D. Spiteller, T. Mironenko, T. L. Li, D. O'Hagan, P. F. Leadlay and J. B. Spencer, Chem. Biol., 2006, 13, 475–484 CrossRef CAS
- A. S. Eustáquio and B. S. Moore, Angew. Chem. Int. Ed. Engl., 2008, 47, 3936–3938 CrossRef CAS
- Y. A. Chan, M. T. Boyne II, A. M. Podevels, A. K. Klimowicz, J. Handelsman, N. L. Kelleher and M. G. Thomas, Proc. Natl. Acad. Sci USA, 2006, 103, 14349–14354 CrossRef CAS
- Y. Sun, H. Hong, F. Gillies, J. B. Spencer and P. F. Leadlay, ChemBioChem, 2008, 9, 150–156 CrossRef CAS
- T.-W. Yu, L. Bai, D. Clade, D. Hoffmann, S. Toelzer, K. Q. Trinh, J. Xu, S. J. Moss, E. Leistner and H. G. Floss, Proc. Natl. Acad. Sci USA, 2002, 99, 7968–7973 CrossRef CAS
- T. Schuhmann, D. Vollmar and S. Grond, J. Antibiot., 2007, 60, 52–60 CrossRef CAS
- A. Rascher, Z. Hu, N. Viswanathan, A. Schirmer, R. Reid, W. C. Nierman, M. Lewis and C. R. Hutchinson, FEMS Microbiol. Lett., 2003, 218, 223–300 CrossRef CAS
- A. Rascher, Z. Hu, G. O. Buchanan, R. Reid and C. R. Hutchinson, Appl. Environ. Microbiol., 2005, 71, 4862–4871 CrossRef CAS
- S. Ōmura, K. Tsuzuki, A. Nakagawa and G. Lukacs, J. Antibiot., 1983, 36, 611–613 CAS
- E. Rodriguez, S. Ward, H. Fu, W. P. Revill, R. McDaniel and L. Katz, Appl. Genet. Mol. Biotechnol., 2004, 66, 85–91 Search PubMed
- S. J. Kakavas, L. Katz and D. Stassi, J. Bacteriol., 1997, 179, 7515–7522 CAS
- C. Zhao, J. Ju, S. D. Christenson, W. C. Smith, D. Song, X. Zhou, B. Shen and Z. Deng, J. Bacteriol., 2006, 188, 4142–4147 CrossRef CAS
- J. Ligon, S. Hill, J. Beck, R. Zirkle, I. Molnár, J. Zawodny, S. Money and T. Schupp, Gene, 2002, 285, 257–267 CrossRef CAS
- W. Li, J. Ju, H. Osada and B. Shen, J. Bacteriol., 2006, 188, 4148–4152 CrossRef CAS
- K. M. Byrne, A. Shafiee, J. B. Nielsen, B. Arison, R. L. Monaghan and L. Kaplan, Dev. Indust. Microbiol., 1993, 32, 29–45 Search PubMed
- C. D. Reeves, L. M. Chung, Y. Liu, Q. Xue, J. R. Carney, W. P. Revill and L. Katz, J. Biol. Chem., 2002, 277, 9155–9159 CrossRef CAS
- S. C. Wenzel, R. M. Williamson, C. Grünanger, J. Xu, K. Gerth, R. A. Martinez, S. J. Moss, B. J. Carroll, S. Grond, C. J. Unkefer, R. Müller and H. G. Floss, J. Am. Chem. Soc., 2006, 128, 14325–14336 CrossRef CAS
- T. Schuhmann and S. Grond, J. Antibiot., 2004, 57, 655–661 CAS
- A. Haber, R. D. Johnson and K. L. Rinehart, Jr., J. Am. Chem. Soc., 1977, 99, 3541–3544 CrossRef CAS
- S. Ōmura, A. Nakagawa, H. Takeshima, K. Atusmi and J. Miyazawa, J. Am. Chem. Soc., 1975, 97, 6600–6602 CrossRef CAS
- K. Hatano, E. Mizuta, S. Akiyama, E. Higashide and Y. Nakao, Agricult. Biol. Chem., 1985, 49, 327–333 Search PubMed
- A. M. Hill, J. P. Harris and A. P. Siskos, Chem. Commun., 1998, 21, 2361–2362 Search PubMed
- B. J. Carroll, S. J. Moss, L. Bai, Y. Kato, S. Toelzer, T.-W. Yu and H. G. Floss, J. Am. Chem. Soc., 2002, 124, 4176–4177 CrossRef CAS
- S. Yue, J. S. Duncan, Y. Yamamoto and C. R. Hutchinson, J. Am. Chem. Soc., 1987, 109, 1253–1255 CrossRef CAS
- D. E. Cane and C.-C. Yang, J. Am. Chem. Soc., 1987, 109, 1255–1257 CrossRef CAS
- T.-W. Yu, L. Bai, D. Clade, D. Hoffman, S. Toelzer, K. Q. Trinh, J. Xu, S. J. Moss, E. Leistner and H. G. Floss, Proc. Natl. Acad. Sci. USA, 2002, 99, 7968–7973 CrossRef CAS
- Y. Kato, L. Bai, Q. Xue, W. P. Revill, T.-W. Yu and H. G. Floss, J. Am. Chem. Soc., 2002, 124, 5268–5269 CrossRef CAS
- K. Watanabe, C. Khosla, R. M. Stroud and S.-C. Tsai, J. Mol. Biol., 2003, 334, 435–444 CrossRef CAS
- M. A. Rude and C. Khosla, J. Antibiot., 2006, 59, 464–470 CrossRef CAS
- L. J. Walton, C. Corre and G. L. Challis, J. Ind. Microbiol. Biotechnol., 2006, 33, 105–120 CrossRef CAS
- M. Ono, S. Sakuda, H. Ikeda, K. Furihata, J. Nakayama, A. Suzuki and A. Isogai, J. Antibiot., 1998, 51, 1019–1028 CAS
- S. Sakuda, M. Ono, H. Ikeda, Y. Inagaki, J. Nakayama, A. Suzuki and A. Isogai, Tetrahedron Lett., 1997, 38, 7399–7402 CrossRef CAS
- L. A. Siloh-Suh, B. J. Lethbridge, S. J. Raffel, H. He, J. Clardy and J. Handelsman, Appl. Environ. Microbiol., 1994, 60, 2023–2030
- E. A. B. Emmert, A. K. Klimowicz, M. G. Thomas and J. Handelsman, Appl. Environ. Microbiol., 2004, 70, 104–113 CrossRef CAS
- J. Shoji, R. Sakazaki, Y. Wakisaka, K. Koizumi, M. Mayama and S. Matsuura, J. Antibiot., 1975, 28, 122–125 CAS
- K. Gebhardt, J. Schimana, J. Müller, H.-P. Fiedler, H. G. Kallenborn, M. Holzenkämpfer, P. Krastel, A. Zeeck, J. Vater, A. Höltzel, D. G. Schmid, J. Rheinheimer and K. Dettner, FEMS Microbiol. Lett., 2002, 217, 199–205 CrossRef CAS
- E. Sarubbi, F. Monti, E. Corti, A. Miele and E. Selva, Eur. J. Biochem., 2004, 271, 3146–3154 CrossRef CAS
- J. J. Lee, J. P. Lee, P. J. Keller, C. E. Cottrell, C.-J. Chang, H. Zähner and H. G. Floss, J. Antibiot., 1986, 39, 1123–1134 CAS
- H.-R. Park, K. Furihata, Y. Hayakawa and K. Shin-ya, Tetrahedron Lett., 2002, 43, 6941–6945 CrossRef CAS
- J. M. Bulsing, E. D. Laue, F. J. Leeper, J. Staunton, D. H. Davies, G. A. F. Ritchie, A. Davies, A. B. Davies and R. P. Mabelis, Chem. Commun., 1984, 1301–1302 RSC
- T. Tamaoki and F. Tomita, J. Antibiot., 1983, 36, 595–598 CAS
- X.-Y. Jia, Z.-H. Tian, L. Shao, X.-D. Qu, Q.-F. Zhao, J. Tang, G.-L. Tang and W. Liu, Chem. Biol., 2006, 13, 575–585 CrossRef CAS
- H. Zhang, J. A. White-Phillip, C. E. Melançon, 3rd, H. J. Kwon, W. L. Yu and H. W. Liu, J. Am. Chem. Soc., 2007, 129, 14670–14683 CrossRef CAS
- Y. Demydchuk, Y. Sun, H. Hong, J. Staunton, J. B. Spencer and P. F. Leadlay, ChemBioChem, 2008, 9, 1136–1145 CrossRef CAS
- H. Motamedi and A. Shafiee, Eur. J. Biochem., 1998, 256, 528–534 CrossRef CAS
- K. Umezawa, Y. Ikeda, O. Kawase, H. Naganawa and S. Kondo, J. Chem. Soc. Perkin Trans. I, 2001, 1550–1553 RSC
| This journal is © The Royal Society of Chemistry 2009 |