Recent developments in ruthenium anticancer drugs
Aviva
Levina
,
Anannya
Mitra
and
Peter A.
Lay
*
School of Chemistry, The University of Sydney, Sydney NSW 2006, Australia. E-mail: p.lay@chem.usyd.edu.au; Fax: +61-2-93513329; Tel: +61-2-93514269
First published on 13th August 2009
Abstract
Interest in Ru anticancer drugs has been growing rapidly since NAMI-A ((ImH+)[RuIIICl4(Im)(S-dmso)], where Im = imidazole and S-dmso = S-bound dimethylsulfoxide) or KP1019 ((IndH+)[RuIIICl4(Ind)2], where Ind = indazole) have successfully completed phase I clinical trials and an array of other Ru complexes have shown promise for future development. Herein, the recent literature is reviewed critically to ascertain likely mechanisms of action of Ru-based anticancer drugs, with the emphasis on their reactions with biological media. The most likely interactions of Ru complexes are with: (i) albumin and transferrin in blood plasma, the former serving as a Ru depot, and the latter possibly providing active transport of Ru into cells; (ii) collagens of the extracellular matrix and actins on the cell surface, which are likely to be involved in the specific anti-metastatic action of Ru complexes; (iii) regulatory enzymes within the cell membrane and/or in the cytoplasm; and (iv) DNA in the cell nucleus. Some types of Ru complexes can also promote the intracellular formation of free radical species, either through irradiation (photodynamic therapy), or through reactions with cellular reductants. The metabolic pathways involve competition among reduction, aquation, and hydrolysis in the extracellular medium; binding to transport proteins, the extracellular matrix, and cell-surface biomolecules; and diffusion into cells; with the extent to which individual drugs participate in various steps along these pathways being crucial factors in determining whether they are mainly anti-metastatic or cytotoxic. This diversity of modes of action of Ru anticancer drugs is also likely to enhance their anticancer activities and to reduce the potential for them to develop tumour resistance. New approaches to metabolic studies, such as X-ray absorption spectroscopy and X-ray fluorescence microscopy, are required to provide further mechanistic insights, which could lead to the rational design of improved Ru anticancer drugs.
 Aviva Levina | Dr Aviva Levina is a Senior Research Associate with Prof. Peter A. Lay at the School of Chemistry, The University of Sydney. She received her PhD from Riga Technical University, Latvia, in 1992. After a postdoctoral fellowship with Dr Jacques Muzart at the University of Reims, France (as a French Government scholar), she joined Prof. Lay’s group in 1995. Her work involves various aspects of bioinorganic chemistry, particularly anti-diabetic and genotoxic activities of chromium complexes, and biological applications of X-ray absorption spectroscopy. |
 Anannya Mitra | Dr Anannya Mitra is a visiting fellow with Prof. Peter A. Lay at the School of Chemistry, The University of Sydney. She received her PhD from Jadavpur University, India, in 2004, as a CSIR fellow. She continued her postdoctoral research at CSIR Durgapur, India, in collaboration with Prof. Dr (mult) Rudi van Eldik, Erlangen University, Germany, before joining Prof Lay’s group in January 2007. She is engaged in studying the kinetics and mechanism of the interaction of platinum group metals with DNA-related molecules and other biomolecules, using conventional and fast kinetic stopped-flow spectrophotometric techniques, and identification of reaction intermediates with X-ray absorption spectroscopy. |
 Peter A. Lay | Professor Lay BSc(Hon 1) (UMelb, 1977), PhD (ANU, 1981), FAA, was a CSIRO Postdoctoral Fellow (1981-1984) at Stanford University and CSIRO, and a QEII Fellow (1984–1985) at Deakin University and the ANU. In 1985, he became a Lecturer in Inorganic Chemistry at The University of Sydney, where he now holds a personal chair (1997–present) and was awarded Australian Research Council Professorial Fellowships for 2002–2007 and 2009–2013. Professor Lay has received the Rennie, Burrows and H. G. Smith Medals (Royal Australian Chemical Institute) for his research in inorganic and bioinorganic chemistry, including metal-containing pharmaceuticals, which has made extensive use of synchrotron science. |
Cisplatin and its derivatives: a blueprint for the development of ruthenium anticancer drugs
A steady growth of interest in Ru anticancer drugs over the last 20 years is reflected in the accelerating growth of publications in this area (Fig. 1, based on a search in the Chemical Abstracts database).1 The biological activities (including anticancer activities) of Ru complexes were first recognised by Dwyer and co-workers in the 1950s,2–4 but this research was largely forgotten until the serendipitous discovery of cisplatin by Rosenberg and co-workers in the 1960s,5–7 and the subsequent developments in Ru anticancer drugs often mirrored those of Pt drugs. Hence, it is logical to start with a brief overview of the currently accepted mechanism of anticancer activity of Pt complexes (Scheme 1).6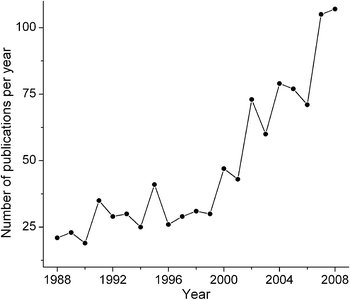 | ||
| Fig. 1 Growth in publications on Ru anticancer drugs (searched in the Chemical Abstracts database).1 | ||
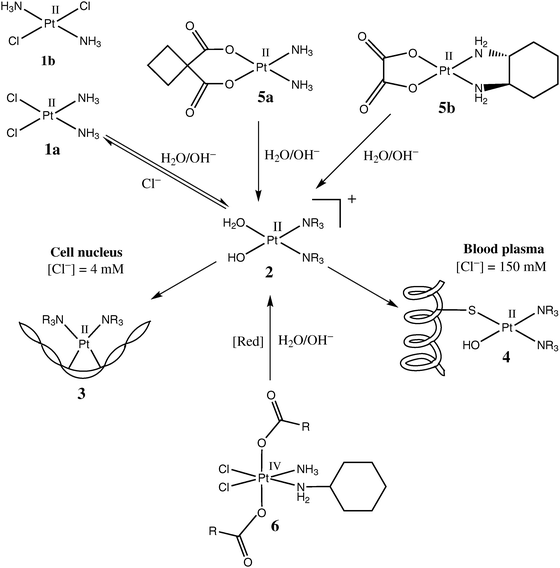 | ||
| Scheme 1 Commonly accepted mechanism of action of Pt anticancer drugs (based on the data from ref. 6). | ||
The cytotoxicity of cisplatin (cis-[PtIICl2(NH3)2], 1a in Scheme 1) in common cancer cell lines is several orders of magnitude higher than that of transplatin (1b in Scheme 1),8 which shows that isomers with the same donor ligands can have quite different biological activities. Under physiological conditions, the relatively labile chlorido ligands (also called leaving groups) of 1a are replaced within several hours by aqua/hydroxido ligands, which leads to partially hydrolysed species such as 2 (Scheme 1), which then readily bind to biological macromolecules.9 It is thought that in order to execute its cytotoxic function, a molecule of 1a has to diffuse unchanged into the cell nucleus, where aquation and hydrolysis would occur, followed by DNA binding (3 in Scheme 1).6,7 In some cases, binding of an aquation and/or hydrolysis product of 1a to two adjacent guanine bases in the DNA chain can cause a severe distortion in the DNA structure (as illustrated in 3), which is not repaired by the existing enzymatic mechanisms, and triggers cell deathviaapoptosis.6,7 Premature aquation and hydrolysis of 1a in the extracellular medium leads to the formation of Pt adducts with proteins (primarily serum albumin , 4 in Scheme 1).10 This undesirable process is thought to deactivate up to 98% of 1a administered by intravenous injection, despite the fact that the rate of aquation of 1a in the blood plasma is lower than that in the cells due to the higher concentration of Cl− ions (150 mM extracellularversus 4–20 mM intracellular),6,7,11i.e., Cl− competes more effectively as a nucleophile than does H2O or proteins at the higher extracellular concentration of Cl−. Further deactivation of 1a can occur in the cytoplasm due to its binding to intracellularS-donors, such as glutathione (binding of Pt(II) to S-donors is favoured due to the “soft” nature of both Pt and S atoms).6,11 It has to be emphasised, however, that alternative cytotoxicity mechanisms to that outlined in Scheme 1 are now seriously considered as contributing to the biological activities of these drugs. These mechanisms include the interactions of Pt complexes with the plasma membrane,12 or with regulatory proteins.13 Generally, the rates of ligand-exchange reactions that are comparable with those of cell-division processes seem to be responsible for the anticancer activity of Pt(II) complexes.10
While Pt drugs are used extensively in oncology, there is a limited range of activity of cisplatin (mainly used in the treatment of testicular and ovarian cancers), although it has a much wider range of activity in combination therapy.14 The main issues with cisplatin, apart from its limited range of activity, are its high systemic toxicity15 and the propensity for patients to develop tumour resistance,16 all of which have led to considerable efforts to design alternative Pt anticancer drugs with lower toxicity, fewer issues with tumour resistance, and/or a broader spectrum of activity.14,17 As a result of synthesis and testing of thousands of drug candidates, only two more Pt(II) complexes (carboplatin and oxaliplatin, 5a and 5b in Scheme 1) have reached wide clinical use.6,7,18 The use of carboxylato instead of chlorido ligands as leaving groups (5a and 5b), and the use of a more inert chelating N-donor ligands instead of NH3 (5b) cause an increase in water solubility and favourable changes in the ligand-exchange reaction rates compared with 1a.14,17,19 Much recent effort has also been directed in the last decade towards the design of orally active Pt(IV) compounds, such as satraplatin (6 in Scheme 1), but all these compounds are still in the pre-clinical research stage.18–20 In contrast to square-planar Pt(II) complexes, 1a,b or 5a,b, octahedral Pt(IV) compounds, such as 6, are stable for days in biological media, so that they can reach the tumour site unchanged, and then be gradually converted to more labile Pt(II) species by biological reductants (such as ascorbate, glutathione and NAD(P)H), assisted by the hypoxic environment that typically exists within solid tumours.20 The accumulated experience in the development of platinum anticancer drugs provided the impetus to explore the more versatile ligand-exchange and redox properties of other Pt-group metals, including Ru.14,21,22
Structures and activities of ruthenium anticancer drugs
Studies of medicinal applications of Ru compounds have been facilitated by the wide diversity of the coordination and organometallic chemistry of Ru that has been developed in fundamental research,23,24 as well as in relation to the use of complexes of this element in catalysis,25 and in photochemistry.26 While Ru(III) is the predominant oxidation state under physiological conditions, Ru(II) and Ru(IV) oxidation states are readily accessible in the presence of biological reductants (e.g., ascorbate or glutathione) or oxidants (O2 or H2O2), respectively.19,27,28 Even Ru(V) has been postulated to be involved,27 but the evidence for this is less certain. All three of these oxidation states (Ru(II), Ru(III) and Ru(IV)) typically form octahedral coordination compounds (mostly with relatively “soft” nitrogen and sulfur donor ligands), while typical Ru(II) organometallic complexes are tetrahedral (piano-stool pseudo-octahedral geometry), including at least one π-bond to an arene ligand.19,25,26,29The history of development of Ru anticancer drugs has been extensively reviewed.19,21,28–35 Briefly, one of the earliest types of anticancer Ru complexes, proposed by Clarke and co-workers in the 1980s,30,36,37 were the chlorido-ammine Ru(II) and Ru(III) complexes, such as 7 in Chart 1, clearly inspired by cisplatin (1a in Scheme 1), and which were thought to act primarily by binding to DNA.30 Among other Ru–ammine complexes tested for their cytotoxicity in cancer cell lines 30,37 was a well-known cytological dye, ruthenium red (8 in Chart 1), which is thought to inhibit Ca(II) transport into cells by selective binding to Ca(II)-transporting proteins.30,37,38
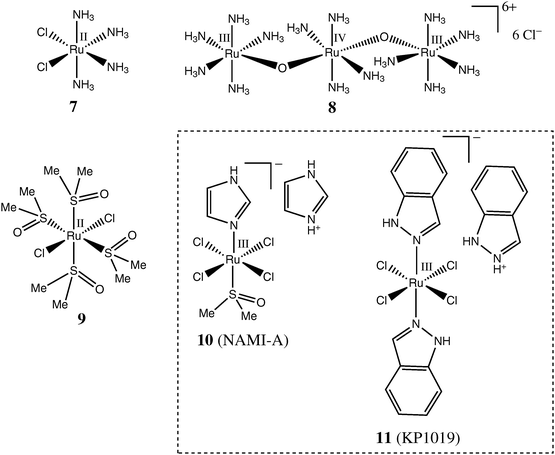 | ||
| Chart 1 Representative Ru complexes with monodentate ligands tested as anticancer drugs. | ||
One of the main issues with biological applications of uncharged complexes, such as 1a or 7, is their poor water solubility.29 Therefore, the next major class of compounds to be studied (by Alessio, Sava and co-workers) were the highly water-soluble Ru(II) chlorido-dmso complexes (where dmso is an S-bound dimethylsulfoxide ligand).39–41 It was found that the trans-[RuCl2(dmso)4] complex (9 in Chart 1) was much more cytotoxic than its cis-counterpart.39,42 This is in stark contrast to the relative activities of the geometric isomers of Pt(II) complexes 1a and 1b (Scheme 1),8 which pointed to likely differences in the mechanisms of action of Ru(II) and Pt(II) complexes.42 Furthermore, it was established that some Ru(II) chlorido-dmso complexes possessed anti-metastatic activity (particularly in non-small cell lung cancer) while being relatively inactive against primary tumours.40,43 Recent developments in this area include the syntheses and biological testing of Ru(II)–dmso complexes with chelating ligands,41,44 as well as of Os(II)–dmso complexes.45 The introduction of chelating ligands generally increases the stability of Ru(II)–dmso complexes against aquation and hydrolysis in aqueous solutions, but no obvious correlation between the aquation rates and biological activities has been found.41
The search for more biologically active compounds related to 9 led to the development of NAMI-A (10 in Chart 1), a specifically anti-metastatic drug that has recently completed phase 1 clinical trials.29,32,46 Since 10 undergoes aquation and hydrolysis within minutes in aqueous media at pH = 7.4 and 37 °C47 and is practically non-cytotoxic in common cancer cell lines ,48 it is clear that its activity is not related to DNA binding in the cell nucleus.32,41 Interactions with actin-type proteins on the cell surface,49,50 or with collagens of the extracellular matrix,51,52 which lead to reduced mobility of invasive cancer cells, have been suggested as possible mechanisms of anti-metastatic action of 10. Numerous analogues of NAMI-A (including Os(III) complexes) have been synthesized and characterised,32,45,48,53,54 but no major improvements in their anti-metastatic activity compared with the parent drug have been reported as yet.
Contemporary with the development of NAMI-A by Sava and co-workers, Keppler and co-workers discovered a Ru(III) chlorido-indazole complex, KP1019 (11 in Chart 1). The KP1019drug is more stable toward aquation and hydrolysis and is more readily taken up by cells than is NAMI-A;53,55,56 it also shows a remarkable activity against primary cisplatin-resistant colorectal tumours, but no pronounced anti-metastatic activity.28,57 This drug has also recently completed phase 1 clinical trials.58,59 Since both NAMI-A (10) and KP1019 (11) complexes readily react with biological reductants (e.g., ascorbate or glutathione) in protein-free model systems,60–62 it was suggested that the reduction of Ru(III) pro-drugs to Ru(II) species may be required for their biological activity, as in the case of Pt(IV) complexes, such as 6 in Scheme 1 (activation by reduction hypothesis).28 Although experimental evidence points to an increase in anti-metastatic activity of 10 in the presence of biological reductants,60 no direct studies of the changes of Ru oxidation states in cells and tissues (similar to those of Pt(IV) conversion to Pt(II))63 have been reported as yet.
The most numerous group of cytotoxic Ru compounds are Ru(II) arene complexes, which were developed primarily by Dyson and co-workers19,34,64 and Sadler and co-workers,11,33,65 although none of these compounds has yet entered clinical trials. One of the motivations for the development of these air-stable Ru(II) complexes was the activation by reduction hypothesis, which suggested that active Ru(II) species may be formed in vivo from Ru(III) precursors, such as 10 or 11.11,28 Typical Ru(II)–arene complexes, such as 12 (RAPTA-C)64 and 1365 in Chart 2 possess Cl− leaving groups and aquate and hydrolyse readily at pH = 7.4 and low Cl− concentrations (corresponding to intracellular conditions), but the aquation equilibrium and rate are strongly inhibited by excess Cl− (corresponding to the blood plasma conditions), similarly to the conversion of 1a into 2 in Scheme 1.66,67 The cytotoxicities of 12, 13 and related compounds are thought to be caused by the binding of the aquation products to DNA, in a similar manner as described for 3 in Scheme 1.19,33 The kinetics of aquation and cellular uptake of Ru(II) arene complexes can be fine-tuned by changing the nature of the metal centre (Ru(II) or Os(II)) or of the arene ligand, or by introducing a chelating ligand (as illustrated by the structures 12 and 13 in Chart 2);68–72 detailed surveys of such compounds can be found in recent reviews.11,19,33,34,64,65,73 As a result of the comprehensive nature of these recent reviews, it is not necessary to cover these areas in as much depth as the other areas covered in the current review, but this in no way diminishes the importance of these interesting new classes of drugs, which may emerge as being clinically significant.
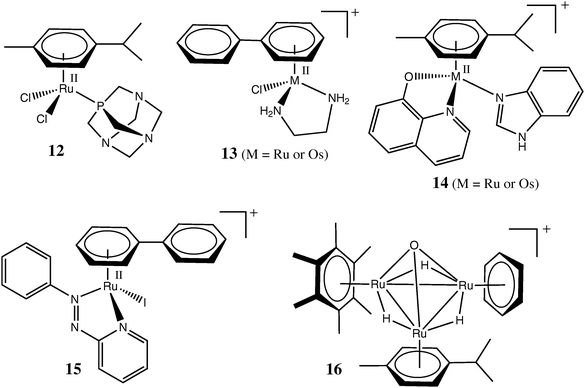 | ||
| Chart 2 Representative organometallic Ru or Os complexes tested as anticancer drugs. | ||
Some of the recently synthesized Ru(II) arene complexes, such as 14 and 15 (Chart 2)74,75 are notable for their relatively high solubility in water combined with very high stability towards aquation. It is thought that 15 is taken up unchanged into the cytoplasm, where it acts as a catalyst of glutathioneoxidation by O2, which leads to an increase in cellular oxidative stress and promotes cell deathviaapoptosis.75 Notably, the catalysis is thought to be promoted by the nucleophilic addition of the thiolato group of glutathione to the azo group of the ligand, rather than to the Ru(II) centre, in 15.75 This unusual mechanism may also contribute to the cytotoxicity of the [RuII(azpy)2Cl2] or [RuII(azpy)3]2+ type complexes (where azpy are the arylazopyridine ligands similar to that in 15), which were studied in detail by Reedijk and co-workers.76,77 Another recently emerged function of Ru(II)–arene complexes is the inhibition of several types of enzymes thought to be involved in cancer progression (thioredoxin reductase and catpepsin B)78 or in the development of resistance to anticancer drugs (glutathione S-transferrase).79 In both cases, a Ru complex is likely to bind to a cysteine or selenocysteine residue in the active site of an enzyme.78,79 Finally, some RAPTA-type complexes (analogues of 12) have recently been shown to possess anti-metastatic activity similar to that of 10.80
One of the most remarkable recent findings is the high cytotoxicity of trinuclear Ru–arene clusters, such as 16 in Chart 2 (and its analogues with various arene ligands) in ovarian cancer cell lines , while the cytotoxicity of closely related tetranuclear Ru clusters under the same conditions was at least an order of magnitude lower.81 An example of a supramolecular chemistry approach to the development of novel metal-based anti-cancer drugs involves the synthesis of cage-like polynuclear Ru(II) arene complexes that encapsulate cytotoxic Pt(II) complexes. This approach is postulated to lead to synergistic actions of both classes of complexes.82 Binding of Ru(III/II) complexes to antibodies, or lipid-based nanovectors, for targeted delivery to cancer cells has been proposed.83,84 Recently, the first example of a 106Ru-radiolabelled arene complex, which can be used in the studies of metabolism of Ru anticancer drugs and in tumour imaging, has also been reported.85 This radioisotope has also been used for a long time in the clinic in implants of metallic Ru enriched with 106Ru isotope (half life ∼1 year) for radiotherapy of ophthalmological cancers.86
Representative examples of Ru(III/II) complexes with chelating ligands, tested as potential anticancer drugs, are shown in Chart 3. The chemistry of Ru(III) polyaminecarboxylato complexes (such as 17 in Chart 2) has been extensively studied,27,87 and they exhibit considerable cytotoxicity against common cancer cell lines .88,89 These complexes are characterised by unusually rapid exchange of chlorido ligands for aqua ligands or for donor groups of biomolecules. They also have a propensity for oxidation to Ru(IV) (and possibly Ru(V)) species under biologically relevant conditions (by aerial dioxygen or H2O2 at pH = 5–8).87,90 Therefore, likely modes of anticancer action of these complexes include rapid ligand-exchange reactions with active centres of enzymes (such as cysteine residues of proteases or protein tyrosine phosphatases), as well as induction of cellular oxidative stress.27,91,92 In addition, 17 and its analogues can affect cellular signalling by acting as NO scavengers.27
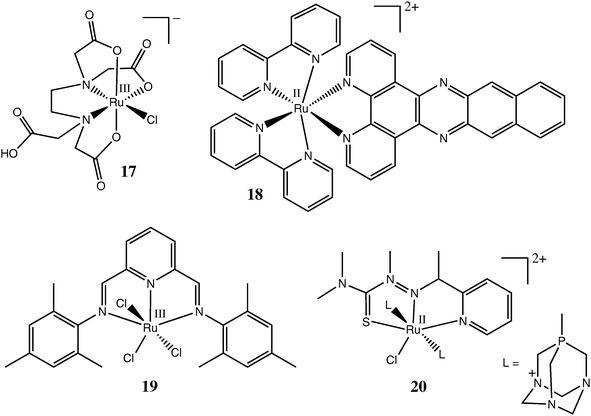 | ||
| Chart 3 Representative Ru complexes with polydentate ligands tested as anticancer drugs. | ||
Well-known Ru(II) complexes with polypyridyl ligands26 have been extensively tested for their DNA-binding and cytotoxic properties, starting from the work of Dwyer and co-workers in the 1960s.3,30,93,94 For instance, the in vitro cytotoxicity of complex 18 (Chart 3) in human colon and breast cancer cell lines was comparable to that of cisplatin.95 Unlike the complexes mentioned previously, 18 does not have any potential leaving groups, and probably acts as a DNAintercalator, due to the presence of a large aromatic polycyclic ligand.93 Recently, other types of polydentate N-donor ligands, such as bis(arylimino)pyridine in 19 (Chart 3), have been proposed as more easily accessible and versatile alternatives to polypyridyls for the synthesis and biological screening of Ru(III/II) complexes.96 A water-soluble Ru(II) complex with a tridentate thiosemicarbazone ligand, 20 (Chart 3), showed higher cytotoxicity towards cancer cell lines in slightly acidic media (pH = 6.0) compared with neutral media (pH = 7.4), which may be beneficial for its selective action in tumour tissues that often have somewhat higher acidity compared with the surrounding normal tissues.97
The well-known photosensitivity of Ru(II) complexes with polypyridyl or macrocyclic ligands26 prompted their testing as sensitizers for photodynamic therapy (PDT) of cancer.98 The Ru complexes suitable for PDT have to conform to the following requirements:30 (i) be both stable towards aquation and relatively non-toxic; (ii) accumulate preferentially in cancer versus normal cells; (iii) absorb strongly at relatively long wavelengths (640–850 nm) that are not significantly absorbed by biological tissues; and (iv) produce high quantum yields of free radical intermediates or singlet oxygen (1O2) on irradiation. A recent example of successful implementation of these principles is a tetranuclear Ru(II)–arene-porphyrin complex 21 (Chart 4) and its analogues, which showed promising activity in PDT of melanoma.99 A special case of PDT is the use of photo-induced NO donors, which lead to massive localised release of NO (a ubiquitous signalling molecule) in cancer cells and trigger their death through apoptosis.100,101 Recent synthesis of Ru(III)–NO complexes, such as 22 (Chart 4), which are able to release NO on irradiation with visible light (∼500 nm) due to the presence of a fluorescent dye as a co-ligand, provide the necessary first step in the development of this type of Ru-based PDT agents.101,102
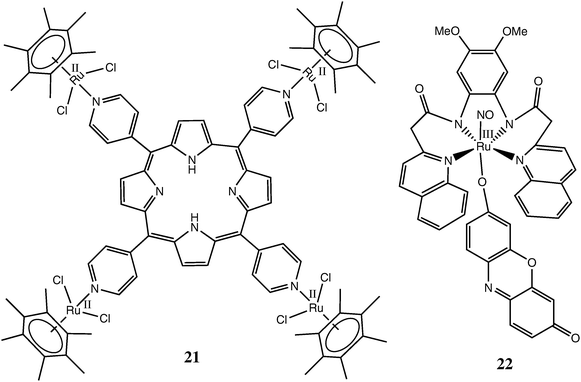 | ||
| Chart 4 Representative Ru complexes tested as active agents for photodynamic therapy. | ||
Complex 18 (Chart 3)95 presents an example of cytotoxicity caused by non-covalent interactions with biomolecules (in this case, DNA intercalation),93 rather than by covalent binding, as shown by 3 and 4 in Scheme 1. Other examples of such interactions are provided by organometallic Ru complexes 23 and 24 (Chart 5),103–105 which have been designed as structural analogues of known organic anticancer drugs (tamoxifen and staurosporine, respectively, see Chart 5). These drugs (and their analogues with other metal ions, such as Fe(II))11 are thought to act by non-covalent binding to specific proteins involved in the development of cancer, such as oestrogen receptors (for tamoxifen)103 or cyclin-dependent kinases (for staurosporine).104 However, whereas Fe-tamoxifen (ferrocifen) is cytotoxic against both ER(+) and ER(−) breast cancer cells, the structurally similar ruthenocene adduct of ferrocifen (23) has antiproliferative action against ER(+), but not against ER(−) breast cancer cells,106 although it is unclear which is more likely to be more active in vivo. In particular, metal complexes like 24 provide a very attractive synthetic alternative to polycyclic systems, such as staurosporine, which are very common among natural biologically active compounds.4 Even more attractive is the use of supramolecular chemistry techniques for self-assembly of elaborate three-dimensional structures, such as a triple-helicate Ru(II) complex 25 (Chart 5).107 The size and shape of complex 25 resembles that of zinc finger proteins, and it shows considerable cytotoxicity in cancer cell lines (much higher than that of a related dinuclear Ru(II) complex with non-helical structure),108 presumably due to the tight non-covalent binding to DNA. Generally, the use of metal (including Ru) ions as scaffolds for the assembly of rigid three-dimensional structures designed for non-covalent interactions with specific biological targets is likely to develop into one of the leading directions in medicinal chemistry.4,21,109
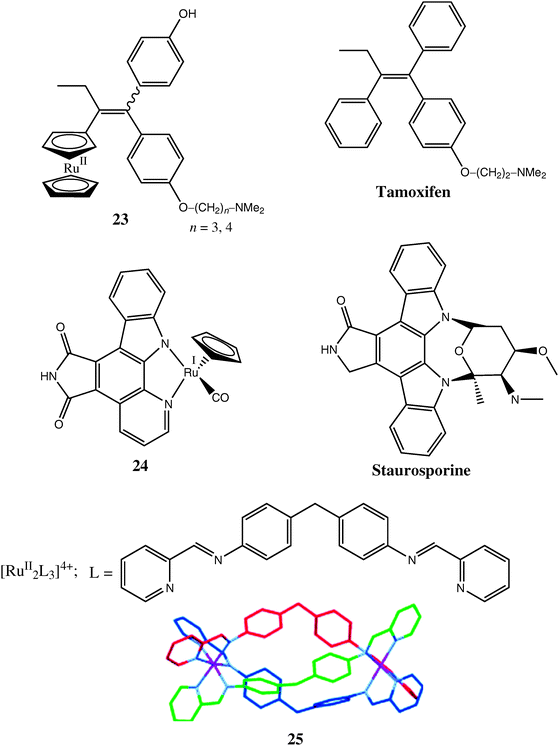 | ||
| Chart 5 Representative Ru complexes designed as structural analogues of known biologically active compounds. | ||
Interactions with biomolecules and general pathways for biological activation of ruthenium anticancer pro-drugs
Numerous reviews on Ru anticancer drugs, published in the last five years, have tended to concentrate either on a single compound, such as NAMI-A (10) 29,31,32 or KP1019 (11),28,58,110 or on a group of complexes with similar ligands, such as ammine,30 polypyridyls,30,93,94 polyaminecarboxylates,27 or arenes,19,33,34,65,73 and their different modes of reactivity in biological media. On the other hand, very recent evidence in the literature points to similarities in the mechanisms of action of various types of Ru anticancer complexes. For instance, specific anti-metastatic activity, which was until recently regarded as a unique property of NAMI-A and its close analogues, has also been demonstrated for RAPTA-type Ru(II)–arene complexes.80 As with NAMI-A, the anti-metastatic activity of Ru(II)–arene complexes appears to be related to interactions with the extracellular matrix and the cell surface, rather than with DNA in the cell nucleus.80 Another example is a common mechanism of enzyme inhibition, based on binding of the Ru ion to S- or Se-donor groups in the active sites of enzymes that has been proposed for both Ru(III)–polyaminecarboxylato91,92 and Ru(II)–arene78,79 complexes.In this section, we bring together what is known about the chemistry and biochemistry of the diverse range of Ru drugs to hypothesise whether all of the activities of the Ru drugs can be described within the constraints of common chemical pathways for the reactions in biological fluids, extracellular matrices, cell surfaces, and intracellular targets. This hypothesis attributes the different biological activities of the Ru complexes to the relative rates by which the Ru complexes react with various biomolecules within or outside the cells, or on the cell surfaces (Scheme 2). At this stage, there are considerable gaps in knowledge about the reactivities of the drugs along these pathways. As such, this review has been designed, in part, to stimulate new research to fill these gaps.
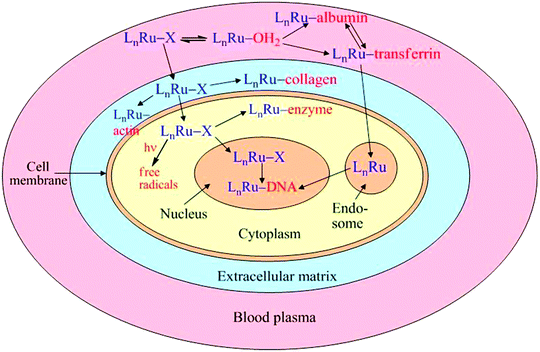 | ||
| Scheme 2 Proposed generalised pathways of action of Ru anticancer drugs (L are the tightly bound ligands and X is the leaving group). | ||
From the data presented in the previous sections, it is clear that interactions with biological macromolecules are crucial for both activities and deactivation of Ru anticancer drugs. In the past, most of the studies have concentrated on the binding of Ru complexes to DNA and its components,30,37,93,111,112 while more recently, the main focus has shifted to their interactions with blood plasma proteins.58,113Scheme 2 outlines two main metabolic routes of Ru complexes (designated as LnRu–X in Scheme 2, where L are the strongly bound ligands such as arenes or N-donors, and X are the leaving groups such as chlorido or carboxylato ligands): cell permeability (either by passive diffusion or by specific transport mechanisms) from extracellular to intracellular compartments, and their aquation and binding to biomolecules specific to each compartment.
Intravenously administered Ru complexes can undergo aquation to various extents in blood plasma, followed by their binding to serum proteins, such as albumin or transferrin (Scheme 2).58,113 While the aquation and protein binding reactions may be catalysed by biological reductants, such as ascorbate (through the formation of kinetically labile Ru(II)–Cl intermediates), recent kinetic studies (performed mostly with 11) suggested that reduction to Ru(II) is unlikely to play a major role in the efficient binding of Ru(III) anticancer drugs to blood proteins.58,114 In the case of 11, at least 90% of Ru was bound to albumin (by far the most abundant protein) in the bloodstream, but an equilibrium is thought to exist between Ru species bound to albumin and transferrin (Tf, the main Fe(III) transport protein), so that the former acts as a depot of Ru ions, and the latter might provide their active transport to the cells.28,58
To our knowledge, 11 is the only anticancer Ru complex for which Tf binding has been studied in detail.58,113 Strong binding of Ru(III) from 11 to histidine residues of Tf has been suggested based on the results of circular dichroism (CD) spectroscopy, electrospray mass spectrometry (ESMS) and gel filtration studies,113 as well as on X-ray diffraction data for 11-lactoferrin adducts (lactoferrin is a protein closely related to Tf),115 although it is unclear to what extent Ru(III) binds specifically to the Fe(III) binding sites of Tf. Cellular uptake studies of 11 in the presence of Tf suggested that the highest uptake is achieved when one of the two binding sites of Tf is loaded with Ru(III), and the other one with Fe(III).58 It was proposed that Ru(II) is released from the Ru(III)–Tf complex inside the cells, following its reduction by ascorbate or glutathione.28,58 However, if Ru(III) is indeed taken into cells in a stable complex with Tf, it would follow the metabolic pathway for a Fe(III)–Tf complex, including binding to a Tf receptor on the cell surface, then encapsulation within an endosome (Scheme 2), from which it might be released as a Ru(II) complex (with unspecified biological ligands) by a combination of enzymatic reduction and decrease of pH within an endosome.116 However, given the inert nature of Ru(II) imidazole complexes,117–119 it is unclear whether the postulate that Ru(II) would be released from transferrin under these conditions can be justified. An alternative mechanism of the anticancer activities of Ru(III/II) complexes may involve interference with Fe uptake and metabolism.116 Another metal ion that is thought to owe its anticancer activity to the resemblance of its uptake mechanism to that of Fe(III), namely Ga(III), is known to bind specifically to Tf and to be transported actively into cells. Within the cells, further metabolism of Ga(III) is thought to be blocked due to the inability of Ga(III) to be reduced within endosome, so that Ga(III) is likely to act by starving cancer cells of vital Fe(III).120
Although it has not been discussed in the literature, histidine-bound Ru(III/II) sites in Tf complexes may undergo acid-catalysed N/C linkage isomerisation reactions similar to those observed with a range of imidazole-containing ligands118,119 that would most likely occur in the acidic and reducing environment of endosomes (pH ∼5),116 or hypoxic tumours (pH ∼6).20 This possibility requires further investigation, because of the potential biological significance of such C-bound imidazole linkage isomers in Ru(III/II)–Tf complexes, and imidazole-containing anti-cancer drugs, as a result of the strong labilising effects of C-bound imidazoles on both cis and trans auxiliary ligands. Thus, the presence of even small equilibrium concentrations of such linkage isomers would lead to greatly enhanced reactivity compared with Ru adducts containing only N-bound imidazoles.121 At present, nothing is known about the ability of Ru(III) to follow the Fe(III/II) metabolic pathway after its uptake by cells,58 and more extensive studies of the interactions of Tf with various types of Ru complexes will be required before definitive conclusions can be reached. Another potentially important aspect of reactivity that has not been adequately discussed in the literature and warrants closer scrutiny for the dmso-containing pro-drugs, is S/O-dmso linkage isomerisation reactions, which are known to be oxidation-state dependent.122 Such linkage isomerisation reactions have a marked effect on the redox behaviour and kinetics and thermodynamics of ligand-exchange reactions of imidazole and dmso complexes, and there is a gap in knowledge about the potential importance of these processes in the diverse biological environments encountered by the drugs.
If a Ru complex escapes protein binding in the blood plasma and subsequent excretion, it can diffuse into the extracellular matrix (Scheme 2), and form adducts there with collagens or cell surface proteins (such as actins), which may be responsible for the anti-metastatic action of 1049,50,52 and Ru(II)–arene complexes.80 Any unreacted Ru complex can further diffuse through the cell membrane into the cytoplasm (Scheme 2), and bind to the active centres of various enzymes, either covalently78,79,92 or non-covalently.4,21 Alternatively, Ru complexes can trigger intracellularoxidation reactions, either through activation by visible light (photodynamic therapy, Scheme 2)99 or through depletion of cellular reductants.75 Finally, Ru species that reach the cell nucleus either by diffusion or by active uptake involving the transferrin pathway (Scheme 2), can form DNAadducts , either by covalent binding,112 or non-covalently (by intercalation or by mimicking zinc finger proteins).93,107 The multitude of extracellular and intracellular targets available for interactions with Ru complexes, as illustrated in Scheme 2, may provide a key to the ability of these complexes to overcome tumour resistance,123,124 since cancer cells are less likely to develop a protection mechanism against drugs exerting multiple cytotoxic routes compared with those targeting a particular biochemical pathway.125
According to Scheme 2, the predominant mode(s) of biological action for each Ru complex is(are) likely to be determined by the relative rates of its cellular uptake vs.extracellular aquation, hydrolysis and protein binding. For instance, extracellular interactions are likely to be responsible for the anti-metastatic activity (and lack of significant cytotoxicity) of hydrophilic and labile Ru complexes such as 9 or 10,41,126 while 11, which is more resistant to aquation and is more readily taken into cells, acts primarily as a cytotoxic drug.55Arene complexes of Ru(II) (related to 12), previously evaluated mainly for their cytotoxicity,71,72 have been recently shown to possess anti-metastatic properties similar to those of 10.80 Further studies are required to determine whether the cytotoxic vs. anti-metastatic properties of Ru(II) arene complexes correlate with their aquation and cellular uptake rates. As shown in Scheme 2, the ability to follow the path of a Ru complex from blood plasma to the cell and its compartments, as well as the changes in the chemical state of Ru along this pathway, is crucial for an understanding of the mechanisms of anticancer activity of Ru complexes.
To date, extensive research on the interactions of Ru anticancer drugs (primarily 11) with blood serum proteins58,113 has relied mainly on capillary electrophoresis127–129 or size-exclusion chromatography130 coupled to ICPMS (inductively coupled plasma mass spectrometry, used for determination of Ru). While providing quantitative information about the extent of Ru binding to various types of proteins, these techniques do not detect changes in the oxidation state or coordination environment of Ru (e.g., cleavage of ligands from the original complex). In recent years, a synchrotron-based technique, X-ray absorption spectroscopy (including X-ray near-edge structure or XANES and X-ray absorption fine structure or XAFS components) has developed into a powerful tool for studies of metal speciation in biological systems,92,131–135 such as cellular metabolism of Pt anticancer drugs63,136,137 or binding of 10 to bovine serum albumin .138 In the latter case, a combination of L3- and K-edge spectra of Ru and K-edge spectra of Cl and S was used to cross-reference the changes that occur in the metal centre and in the ligands of 10 upon binding to a protein molecule.138 It was concluded that the metal remains in the Ru(III) oxidation state and the main reaction is Cl− ligand substitution (which may also be the result of aquation of 10 prior to the protein binding, see Scheme 2).138 However, this conclusion is not necessarily valid, since redox catalysis of Ru(III) substitution reactions can occur with amounts of Ru(II) that are too small to be measured by the XANES technique. Such Ru(II) catalysis of substitution of Ru(III) is well-known,139 and chlorido–Ru(II) complexes are very labile to the extent that they could catalyse the substitution reactions with biomolecules via aqua intermediates before reoxidation of the biomolecule Ru(II)-adducts to Ru(III).140
Our group has previously used XANES spectroscopy for detailed studies of metabolism of chromium compounds (including carcinogenic Cr(VI) and purportedly anti-diabetic Cr(III) nutritional supplements) in biological fluids and in cultured cells.141,142 The main advantages of XANES spectroscopy for such studies are the following: (i) specificity toward the studied metal ion, regardless of the physical state and chemical composition of the sample; and (ii) high sensitivity to small changes in the coordination environment of metal ions.143 Multiple linear regression analyses of XANES spectra with the use of a library of model Cr complexes led to assignment of the chemical natures of Cr metabolism products.141,142 Crucially, a comparison of XANES spectra of Cr(III) metabolism products in whole cells and in subcellular fractions pointed to a re-distribution and changes in the coordination environment of Cr(III) caused by the cell lysis and separation procedures.141 These changes mean that metal–protein complexes isolated by chromatographic techniques do not necessarily correspond to the species formed in metal-treated cells, and the use of XANES spectroscopy provides a unique opportunity to delve into changes that take place.141,143
Recently, we applied the same principles to the studies of transformations of typical Ru(III) anticancer drugs, 10 and 11, in cultured mammalian cells, as well as in cell culture media and in blood serum (Z.-J. Lim, Y. Y. Gwee, A. Levina, A. Mitra and P. A. Lay, to be submitted). Unlike our previous studies on Cr141,142 and the published data on Ru,138 we found that the consideration of whole spectra, including XANES and XAFS regions, is essential for the analysis of changes in the coordination environment of Ru compounds in biological media (Fig. 2). For instance, the loss of Cl− ligands during the decomposition of 11 in cell culture medium is manifested in a decrease in the number and intensity of post-edge maxima in the spectra, while changes in the coordination environment of model Ru(III) complexes from N- to O- donor groups cause significant shifts in the positions of these maxima (at 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200–22400 eV, Fig. 2). Multiple linear regression analysis indicated that Ru(III) is bound mainly to N-donors and to a lesser extent to O- and Cl-donors in the decomposition products of 11 (Fig. 2), while gel filtration studies confirmed that the resultant Ru(III) species were fully protein-bound (the protein source was fetal calf serum added to the medium). We expect that further studies in this direction will shed new light onto the fate of Ru anticancer drugs in biological systems.
200–22400 eV, Fig. 2). Multiple linear regression analysis indicated that Ru(III) is bound mainly to N-donors and to a lesser extent to O- and Cl-donors in the decomposition products of 11 (Fig. 2), while gel filtration studies confirmed that the resultant Ru(III) species were fully protein-bound (the protein source was fetal calf serum added to the medium). We expect that further studies in this direction will shed new light onto the fate of Ru anticancer drugs in biological systems.
![Representative K-edge X-ray absorption spectra of model Ru compounds (solids mixed with boron nitride in ∼1 : 10 ratio) and decomposition products of KP1019 in cell culture medium (Advanced DMEM from Invitrogen, supplemented with 2% fetal calf serum and 0.50 mM KP1019, incubated for 4 h at 37 °C, then freeze-dried). The spectra were collected at the Australian National Beamline Facility (beamline 20B at the Photon Factory, Tsukuba, Japan) at 14 K, using the fluorescence detection mode (36-pixel Ge detector). The experimental spectrum of the decomposition products of KP1019 (black line) was fitted (red line, R = 0.9994) with a linear combination of spectra of model compounds: [RuIII(NH3)6]Cl3, 60 ± 2%; [RuIII(acac)3], 14 ± 2% (acac = acetylacetonato = 2,4-pentanedionato(−)); [Ru3IIIO(OAc)6(OH2)3](OAc), 16 ± 3%; and KP1019 (11 in Chart 1), 10 ± 2%. Details of experiments and data processing were similar to those described previously (ref. 141 and 142).](/image/article/2009/MT/b904071d/b904071d-f2.gif) | ||
| Fig. 2 Representative K-edge X-ray absorption spectra of model Ru compounds (solids mixed with boron nitride in ∼1 : 10 ratio) and decomposition products of KP1019 in cell culture medium (Advanced DMEM from Invitrogen, supplemented with 2% fetal calf serum and 0.50 mM KP1019, incubated for 4 h at 37 °C, then freeze-dried). The spectra were collected at the Australian National Beamline Facility (beamline 20B at the Photon Factory, Tsukuba, Japan) at 14 K, using the fluorescence detection mode (36-pixel Ge detector). The experimental spectrum of the decomposition products of KP1019 (black line) was fitted (red line, R = 0.9994) with a linear combination of spectra of model compounds: [RuIII(NH3)6]Cl3, 60 ± 2%; [RuIII(acac)3], 14 ± 2% (acac = acetylacetonato = 2,4-pentanedionato(−)); [Ru3IIIO(OAc)6(OH2)3](OAc), 16 ± 3%; and KP1019 (11 in Chart 1), 10 ± 2%. Details of experiments and data processing were similar to those described previously (ref. 141 and 142). | ||
An even more promising synchrotron-based technique for the study of biotransformations of Ru anticancer drugs is X-ray fluorescence microprobe imaging,144 including studies of Ru distribution in single mammalian cells or in cancer tissues (used previously for Pt anticancer drugs)63,136,145 or in gels after the separation of lysates from metal-treated cells.132,144,146 A combination of microprobe X-ray fluorescence imaging and XANES spectroscopy allows for the determination of the oxidation states and coordination environments of metal ions within single cells and their organelles,92,136,147,148 which is likely to provide crucial information for understanding the mechanisms of action of Ru anticancer drugs, including clarification of the role of transferrin in Ru(III) transport58 and testing of the activation by reduction hypothesis.28 These can be combined with correlative microscopic studies, such as those involving vibrational microprobe images, to examine how the drugs affect the biochemistry of the cells.149,150
Conclusions
For two decades, there was little overlap between the publications of several research groups that have developed different types of Ru anticancer drugs, including NAMI-A,32KP101958 and Ru(II)–arene complexes.33,34 However, recent trends point to the likelihood of common pathways of biological activation for all types of Ru complexes (Scheme 2) that include competing processes of extracellularprotein binding and cellular uptake. Similar to Pt anticancer drugs, the latter process is likely to be responsible for the cytotoxicity of Ru complexes in primary tumours (e.g., for KP1019).58 Unlike Pt complexes, where binding to extracellular proteins is thought to cause the loss of biological activity,10 binding of Ru complexes to extracellular matrix proteins or to the cell surface is likely to be responsible for the anti-metastatic properties of NAMI-A and of some Ru(II)–arene complexes.32,80 The main factors that determine the mode of action of a Ru complex appear to be its lipophilicity (favouring cellular uptake) and the presence of labile ligands such as chlorido or carboxylato (ligands that favour extracellular binding). The possibility of transferrin-mediated uptake of some Ru(III) complexes into cells, followed by intracellular reduction to produce reactive Ru(II) species, has been suggested,28 but is not yet firmly established and is doubtful given the inert nature of Ru(II)–imidazole bonds.118,121 A better understanding of chemical transformations of Ru complexes in biological media is required for the design of compounds with predictable properties, particularly of new types of anti-metastatic drugs. The use of synchrotron-based techniques, such as X-ray absorption spectroscopy and microprobe X-ray fluorescence mapping, is likely to be crucial for this purpose.Acknowledgements
The research has been supported by the Australian Research Council Discovery grants (DP0208409, DP0664706 and DP0984722) to P. A. L., including ARC Professorial Fellowships. X-Ray absorption spectroscopy (XAS) was performed at the Australian National Beamline Facility (Photon Factory, Tsukuba, Japan) with support from the Australian Synchrotron Research Program, which is funded by the Commonwealth of Australia under the Major National Research Facilities program. We thank Prof. M. Nomura (Photon Factory, Tsukuba, Japan), Drs G. J. Foran and J. Hester (ANSTO, Australia), and Dr J. B. Aitken (University of Sydney, Australia) for their help with XAS experiments and Prof. T. W. Hambley for his helpful comments on the manuscript.References
- Chemical Abstracts, American Chemical Society, 2008. http://www.cas.org/aboutcas/index.html.
- F. P. Dwyer, E. C. Gyarfas, W. P. Rogers and J. H. Koch, Nature, 1952, 170, 190–191 CrossRef CAS.
- F. P. Dwyer, E. Mayhew, E. M. F. Roe and A. Shulman, Br. J. Cancer, 1965, 19, 195–199 CAS.
- E. Meggers, Curr. Opin. Chem. Biol., 2007, 11, 287–292 CrossRef.
- B. Rosenberg, L. Van Camp and T. Krigas, Nature, 1965, 205, 698–699 CAS.
- R. A. Alderden, M. D. Hall and T. W. Hambley, J. Chem. Educ., 2006, 83, 728–734 CrossRef CAS.
- J. Reedijk, Eur. J. Inorg. Chem., 2009, 1303–1312 CrossRef CAS.
- M. J. Cleare and J. D. Hoeschele, Platinum Met. Rev., 1973, 17, 2–13 CAS.
- J. Reedijk, Platinum Met. Rev., 2008, 52, 2–11 CrossRef CAS.
- J. Reedijk, Macromol. Symp., 2008, 270, 193–201 CrossRef CAS.
- A. F. A. Peacock and P. J. Sadler, Chem.–Asian J., 2008, 3, 1890–1899 CrossRef CAS.
- A. Rebillard, D. Lagadic-Gossmann and M.-T. Dimanche-Boitrel, Curr. Med. Chem., 2008, 15, 2656–2663 CrossRef CAS.
- D. Sheikh-Hamad, Am. J. Physiol., 2008, 295, F42–F43 CAS.
- S. P. Fricker, Dalton Trans., 2007, 4903–4917 RSC.
- K. Barabas, R. Milner, D. Lurie and C. Adin, Vet. Comp. Oncol., 2008, 6, 1–18 CrossRef CAS.
- P. Borst, S. Rottenberg and J. Jonkers, Cell Cycle, 2008, 7, 1353–1359 Search PubMed.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- S. Kerpel-Fronius, in Analogue-Based Drug Discovery, ed. J. Fischer and G. C. Robin, Wiley-VCH, Weinheim, Germany, 2006, pp. 385–394 Search PubMed.
- W. H. Ang and P. J. Dyson, Eur. J. Inorg. Chem., 2006, 4003–4018 CrossRef CAS.
- M. D. Hall, H. R. Mellor, R. Callaghan and T. W. Hambley, J. Med. Chem., 2007, 50, 3403–3411 CrossRef CAS.
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS.
- M. J. Hannon, Pure Appl. Chem., 2007, 79, 2243–2261 CrossRef CAS.
- M. Schröder and T. A. Stephenson, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon Press, Oxford, 1987, vol. 4, pp. 277–518 Search PubMed.
- C. E. Housecroft, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004, vol. 5, pp. 555–731 Search PubMed.
- M. Wang and C.-J. Li, Top. Organomet. Chem., 2004, 11, 321–336 CAS.
- J. G. Vos and J. M. Kelly, Dalton Trans., 2006, 4869–4883 RSC.
- D. Chatterjee, A. Mitra and G. S. De, Platinum Met. Rev., 2006, 50, 2–12 CrossRef CAS.
- E. Reisner, V. B. Arion, B. K. Keppler and A. J. L. Pombeiro, Inorg. Chim. Acta, 2008, 361, 1569–1583 CrossRef CAS.
- I. Bratsos, S. Jedner, T. Gianferrara and E. Alessio, Chimia, 2007, 61, 692–697 CrossRef CAS.
- M. J. Clarke, Coord. Chem. Rev., 2003, 236, 209–233 CrossRef CAS.
- E. Alessio, G. Mestroni, A. Bergamo and G. Sava, Met. Ions Biol. Syst., 2004, 42, 323–351 CAS.
- A. Bergamo and G. Sava, Dalton Trans., 2007, 1267–1272 RSC.
- S. J. Dougan and P. J. Sadler, Chimia, 2007, 61, 704–715 CrossRef CAS.
- P. J. Dyson, Chimia, 2007, 61, 698–703 CrossRef CAS.
- P. Heffeter, U. Jungwirth, M. Jakupec, C. Hartinger, M. Galanski, L. Elbling, M. Micksche, B. Keppler and W. Berger, Drug Resist. Updates, 2008, 11, 1–16 CrossRef CAS.
- M. J. Clarke, Met. Ions Biol. Syst., 1980, 11, 231–283 CAS.
- M. J. Clarke, Coord. Chem. Rev., 2002, 232, 69–93 CrossRef CAS.
- Y.-J. Chang, H.-L. Kim, S. J. Sacket, K. Kim, M. Han, J.-Y. Jo and D.-S. Im, J. Appl. Pharmacol., 2007, 15, 150–155 Search PubMed.
- E. Alessio, G. Mestroni, G. Nardin, W. M. Attia, M. Calligaris, G. Sava and S. Zorzet, Inorg. Chem., 1988, 27, 4099–4106 CrossRef CAS.
- G. Mestroni, E. Alessio, G. Sava, S. Pacor, M. Coluccia and A. Boccarelli, Met-Based. Drugs, 1994, 1, 41–63 Search PubMed.
- I. Bratsos, A. Bergamo, G. Sava, T. Gianferrara, E. Zangrando and E. Alessio, J. Inorg. Biochem., 2008, 102, 606–617 CrossRef CAS.
- M. Coluccia, G. Sava, F. Loseto, A. Nassi, A. Boccarelli, D. Giordano, E. Alessio and G. Mestroni, Eur. J. Cancer, 1993, 29, 1873–1879 CrossRef.
- G. Sava, S. Pacor, G. Mestroni and E. Alessio, Clin. Exp. Metastasis, 1992, 10, 273–280 CAS.
- V. Mahalingam, N. Chitrapriya, F. R. Fronczek and K. Natarajan, Polyhedron, 2008, 27, 1917–1924 CrossRef CAS.
- A. Egger, B. Cebrian-Losantos, I. N. Stepanenko, A. A. Krokhin, R. Eichinger, M. A. Jakupec, V. B. Arion and B. K. Keppler, Chem. Biodiversity, 2008, 5, 1588–1593 CrossRef CAS.
- G. Sava, I. Capozzi, K. Clerici, G. Gagliardi, E. Alessio and G. Mestroni, Clin. Exp. Metastasis, 1998, 16, 371–379 CrossRef CAS.
- M. Bacac, A. C. G. Hotze, K. van der Schilden, J. G. Haasnoot, S. Pacor, E. Alessio, G. Sava and J. Reedijk, J. Inorg. Biochem., 2004, 98, 402–412 CrossRef CAS.
- A. Bergamo, B. Gava, E. Alessio, G. Mestroni, B. Serli, M. Cocchietto, S. Zorzet and G. Sava, Int. J. Oncol., 2002, 21, 1331–1338 CAS.
- B. Gava, S. Zorzet, P. Spessotto, M. Cocchietto and G. Sava, J. Pharmacol. Exp. Ther., 2006, 317, 284–291 CAS.
- G. Sava, F. Frausin, M. Cocchietto, F. Vita, E. Podda, P. Spessotto, A. Furlani, V. Scarcia and G. Zabucchi, Eur. J. Cancer, 2004, 40, 1383–1396 CrossRef CAS.
- C. Casarsa, M. T. Mischis and G. Sava, J. Inorg. Biochem., 2004, 98, 1648–1654 CrossRef CAS.
- G. Sava, S. Zorzet, C. Turrin, F. Vita, M. Soranzo, G. Zabucchi, M. Cocchietto, A. Bergamo, S. DiGiovine, G. Pezzoni, L. Sartor and S. Garbisa, Clin. Cancer Res., 2003, 9, 1898–1905 CAS.
- M. Groessl, E. Reisner, C. G. Hartinger, R. Eichinger, O. Semenova, A. R. Timerbaev, M. A. Jakupec, V. B. Arion and B. K. Keppler, J. Med. Chem., 2007, 50, 2185–2193 CrossRef CAS.
- B. Cebrian-Losantos, A. A. Krokhin, I. N. Stepanenko, R. Eichinger, M. A. Jakupec, V. B. Arion and B. K. Keppler, Inorg. Chem., 2007, 46, 5023–5033 CrossRef CAS.
- S. Kapitza, M. Pongratz, M. A. Jakupec, P. Heffeter, W. Berger, L. Lackinger, B. K. Keppler and B. Marian, J. Cancer Res. Clin. Oncol., 2005, 131, 101–110 CrossRef CAS.
- A. V. Vargiu, A. Robertazzi, A. Magistrato, P. Ruggerone and P. Carloni, J. Phys. Chem. B, 2008, 112, 4401–4409 CrossRef CAS.
- B. K. Keppler, WO Application 2002059135, Chem. Abstr., 2002, vol. 137, ref. 119658 Search PubMed.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CrossRef CAS.
- F. Lentz, A. Drescher, A. Lindauer, M. Henke, R. A. Hilger, C. G. Hartinger, M. E. Scheulen, C. Dittrich, B. K. Keppler and U. Jaehde, Anti-Cancer Drugs, 2009, 20, 97–103 CrossRef CAS.
- G. Sava, A. Bergamo, S. Zorzet, B. Gava, C. Casarsa, M. Cocchietto, A. Furlani, V. Scarcia, B. Serli, E. Iengo, E. Alessio and G. Mestroni, Eur. J. Cancer, 2002, 38, 427–435 CrossRef CAS.
- P. Schluga, C. G. Hartinger, A. Egger, E. Reisner, M. Galanski, M. A. Jakupec and B. K. Keppler, Dalton Trans., 2006, 1796–1802 RSC.
- M. Brindell, D. Piotrowska, A. A. Shoukry, G. Stochel and R. Eldik, JBIC, J. Biol. Inorg. Chem., 2007, 12, 809–818 CrossRef CAS.
- M. D. Hall, R. A. Alderden, M. Zhang, P. J. Beale, Z. Cai, B. Lai, A. P. J. Stampfl and T. W. Hambley, J. Struct. Biol., 2006, 155, 38–44 CrossRef CAS.
- W. H. Ang, Chimia, 2007, 61, 140–142 CrossRef CAS.
- Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 4764–4776 RSC.
- F. Wang, H. Chen, S. Parsons, I. D. H. Oswald, J. E. Davidson and P. J. Sadler, Chem.–Eur. J., 2003, 9, 5810–5820 CrossRef CAS.
- C. Scolaro, C. G. Hartinger, C. S. Allardyce, B. K. Keppler and P. J. Dyson, J. Inorg. Biochem., 2008, 102, 1743–1748 CrossRef CAS.
- R. Fernandez, M. Melchart, A. Habtemariam, S. Parsons and P. J. Sadler, Chem.–Eur. J., 2004, 10, 5173–5179 CrossRef CAS.
- F. Wang, A. Habtemariam, E. P. L. van der Geer, R. Fernandez, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. A. Fabbiani, P. Lozano-Casal, I. D. H. Oswald, D. I. Jodrell, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269–18274 CrossRef CAS.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS.
- A. Habtemariam, M. Melchart, R. Fernandez, S. Parsons, I. D. H. Oswald, A. Parkin, F. P. A. Fabbiani, J. E. Davidson, A. Dawson, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2006, 49, 6858–6868 CrossRef CAS.
- C. Scolaro, A. B. Chaplin, C. G. Hartinger, A. Bergamo, M. Cocchietto, B. K. Keppler, G. Sava and P. J. Dyson, Dalton Trans., 2007, 5065–5072 RSC.
- M. Melchart and P. J. Sadler, in Bioorganometallics, ed. G. Jaouen, Wiley-VCH, Weinheim, Germany, 2006, pp. 39–64 Search PubMed.
- R. Schuecker, R. O. John, M. A. Jakupec, V. B. Arion and B. K. Keppler, Organometallics, 2008, 27, 6587–6595 CrossRef CAS.
- S. J. Dougan, A. Habtemariam, S. E. McHale, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11628–11633 CrossRef CAS.
- A. C. G. Hotze, M. Bacac, A. H. Velders, B. A. J. Jansen, H. Kooijman, A. L. Spek, J. G. Haasnoot and J. Reedijk, J. Med. Chem., 2003, 46, 1743–1750 CrossRef CAS.
- A. C. G. Hotze, E. P. L. van der Geer, H. Kooijman, A. L. Spek, J. G. Haasnoot and J. Reedijk, Eur. J. Inorg. Chem., 2005, 2648–2657 CrossRef CAS.
- A. Casini, C. Gabbiani, F. Sorrentino, M. P. Rigobello, A. Bindoli, T. J. Geldbach, A. Marrone, N. Re, C. G. Hartinger, P. J. Dyson and L. Messori, J. Med. Chem., 2008, 51, 6773–6781 CrossRef CAS.
- W. H. Ang, A. De Luca, C. Chapuis-Bernasconi, L. Juillerat-Jeanneret, M. Lo Bello and P. J. Dyson, ChemMedChem, 2007, 2, 1799–1806 CrossRef CAS.
- A. Bergamo, A. Masi, P. J. Dyson and G. Sava, Int. J. Oncol., 2008, 33, 1281–1289 CAS.
- B. Therrien, W. H. Ang, F. Cherioux, L. Vieille-Petit, L. Juillerat-Jeanneret, G. Suess-Fink and P. J. Dyson, J. Cluster Sci., 2007, 18, 741–752 CrossRef CAS.
- B. Therrien, G. Suess-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773–3776 CrossRef CAS.
- W. H. Ang, E. Daldini, L. Juillerat-Jeanneret and P. J. Dyson, Inorg. Chem., 2007, 46, 9048–9050 CrossRef CAS.
- M. Vaccaro, R. Del Litto, G. Mangiapia, A. M. Carnerup, G. D'Errico, F. Ruffo and L. Paduano, Chem. Commun., 2009, 1404–1406 RSC.
- J. D. Hoeschele, A. Habtemariam, J. Muir and P. J. Sadler, Dalton Trans., 2007, 4974–4979 RSC.
- H. Abouzeid, R. Moeckli, M.-C. Gaillard, M. Beck-Popovic, A. Pica, L. Zografos, A. Balmer, S. Pampallona and L. Munier Francis, Int. J. Radiat. Oncol., Biol., Phys., 2008, 71, 821–828 CrossRef.
- D. Chatterjee, Coord. Chem. Rev., 1998, 168, 273–293 CrossRef CAS.
- R. Vilaplana, M. A. Romero, M. Quirós, J. M. Salas and F. González-Vilchez, Met-Based. Drugs, 1995, 2, 211–219 Search PubMed.
- S. R. Grguric-Sipka, R. Vilaplana, J. M. Pérez, M. A. Fuertes, C. Alonso, Y. Alvarez, T. J. Sabo and F. González-Vílchez, J. Inorg. Biochem., 2003, 97, 215–220 CrossRef CAS.
- D. Chatterjee, A. Mitra and R. van Eldik, Dalton Trans., 2007, 943–948 RSC.
- D. Chatterjee, A. Sengupta, A. Mitra, S. Basak, R. Bhattacharya and D. Bhattacharya, Inorg. Chim. Acta, 2005, 358, 2960–2965 CrossRef CAS.
- D. Chatterjee, A. Mitra, A. Levina and P. A. Lay, Chem. Commun., 2008, 2864–2866 RSC.
- N. J. Wheate, C. R. Brodie, J. G. Collins, S. Kemp and J. R. Aldrich-Wright, Mini-Rev. Med. Chem., 2007, 7, 627–648 CrossRef CAS.
- E. Corral, A. C. G. Hotze, H. den Dulk, A. Leczkowska, A. Rodger, M. J. Hannon and J. Reedijk, JBIC, J. Biol. Inorg. Chem., 2009, 14, 439–448 CrossRef CAS.
- U. Schatzschneider, J. Niesel, I. Ott, R. Gust, H. Alborzinia and S. Woelfl, ChemMedChem, 2008, 3, 1104–1109 CrossRef CAS.
- A. Garza-Ortiz, P. U. Maheswari, M. Siegler, A. L. Spek and J. Reedijk, Inorg. Chem., 2008, 47, 6964–6973 CrossRef CAS.
- S. Grguric-Sipka, C. R. Kowol, S.-M. Valiahdi, R. Eichinger, M. A. Jakupec, A. Roller, S. Shova, V. B. Arion and B. K. Keppler, Eur. J. Inorg. Chem., 2007, 2870–2878 CrossRef CAS.
- K. Ishii, M. Shiine, Y. Shimizu, S.-i. Hoshino, H. Abe, K. Sogawa and N. Kobayashi, J. Phys. Chem. B, 2008, 112, 3138–3143 CrossRef CAS.
- F. Schmitt, P. Govindaswamy, G. Suess-Fink, W. H. Ang, P. J. Dyson, L. Juillerat-Jeanneret and B. Therrien, J. Med. Chem., 2008, 51, 1811–1816 CrossRef CAS.
- M. J. Rose and P. K. Mascharak, Coord. Chem. Rev., 2008, 252, 2093–2114 CrossRef CAS.
- M. J. Rose, N. L. Fry, R. Marlow, L. Hinck and P. K. Mascharak, J. Am. Chem. Soc., 2008, 130, 8834–8846 CrossRef CAS.
- M. J. Rose and P. K. Mascharak, Chem. Commun., 2008, 3933–3935 RSC.
- A. Vessieres, S. Top, W. Beck, E. Hillard and G. Jaouen, Dalton Trans., 2006, 529–541 RSC.
- J. E. Debreczeni, A. N. Bullock, G. E. Atilla, D. S. Williams, H. Bregman, S. Knapp and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 1580–1585 CrossRef CAS.
- J. Maksimoska, L. Feng, K. Harms, C. Yi, J. Kissil, R. Marmorstein and E. Meggers, J. Am. Chem. Soc., 2008, 130, 15764–15765 CrossRef CAS.
- P. Pigeon, S. Top, A. Vessieres, M. Huche, E. A. Hillard, E. Salomon and G. Jaouen, J. Med. Chem., 2005, 48, 2814–2821 CrossRef CAS.
- G. I. Pascu, A. C. G. Hotze, C. Sanchez-Cano, B. M. Kariuki and M. J. Hannon, Angew. Chem., Int. Ed., 2007, 46, 4374–4378 CrossRef CAS.
- U. McDonnell, J. M. C. A. Kerchoffs, R. P. M. Castineiras, M. R. Hicks, A. C. G. Hotze, M. J. Hannon and A. Rodger, Dalton Trans., 2008, 667–675 RSC.
- G. Jaouen, S. Top and A. Vessieres, in Bioorganometallics, ed. G. Jaouen, Wiley-VCH, Weinheim, Germany, 2006, pp. 65–95 Search PubMed.
- C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbas and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 891–904 CrossRef CAS.
- V. Brabec, Prog. Nucleic Acid Res. Mol. Biol., 2002, 71, 1–68 Search PubMed.
- V. Brabec and O. Novakova, Drug Resist. Updates, 2006, 9, 111–122 CrossRef CAS.
- A. R. Timerbaev, C. G. Hartinger, S. S. Aleksenko and B. Keppler, Chem. Rev., 2006, 106, 2224–2248 CrossRef CAS.
- A. R. Timerbaev, L. S. Foteeva, A. V. Rudnev, J. K. Abramski, K. Polec-Pawlak, C. G. Hartinger, M. Jarosz and B. K. Keppler, Electrophoresis, 2007, 28, 2235–2240 CrossRef CAS.
- C. A. Smith, A. J. Sutherland-Smith, B. K. Keppler, F. Kratz and E. N. Baker, JBIC, J. Biol. Inorg. Chem., 1996, 1, 424–431 CrossRef CAS.
- D. Kalinowski and D. R. Richardson, Pharmacol. Rev., 2005, 57, 547–583 CrossRef CAS.
- D. M. Martin, N. D. Chasteen and J. K. Grady, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1991, 1076, 252–258 Search PubMed.
- R. J. Sundberg, R. F. Bryan, I. F. J. Taylor and H. Taube, J. Am. Chem. Soc., 1974, 96, 381–392 CrossRef CAS.
- R. J. Sundberg and G. Gupta, Bioinorg. Chem., 1973, 3, 39–48 CrossRef CAS.
- N. P. Davies, Y. S. Rahmanto, C. R. Chitambar and D. R. Richardson, J. Pharmacol. Exp. Ther., 2006, 317, 153–162 CAS.
- M. F. Tweedle and H. Taube, Inorg. Chem., 1982, 21, 3361–3371 CrossRef CAS.
- A. Yeh, N. Scott and H. Taube, Inorg. Chem., 1982, 21, 2542–2545 CrossRef CAS.
- P. Heffeter, M. Pongratz, E. Steiner, P. Chiba, M. A. Jakupec, L. Elbling, B. Marian, W. Koerner, F. Sevelda, M. Micksche, B. K. Keppler and W. Berger, J. Pharmacol. Exp. Ther., 2005, 312, 281–289 CAS.
- C. A. Vock, W. H. Ang, C. Scolaro, A. D. Phillips, L. Lagopoulos, L. Juillerat-Jeanneret, G. Sava, R. Scopelliti and P. J. Dyson, J. Med. Chem., 2007, 50, 2166–2175 CrossRef CAS.
- T. W. Hambley, Cancer Res., 2009, 69, 1259–1262 CrossRef CAS.
- S. Zorzet, A. Bergamo, M. Cocchietto, A. Sorc, B. Gava, E. Alessio, E. Iengo and G. Sava, J. Pharmacol. Exp. Ther., 2000, 295, 927–933 CAS.
- K. Polec-Pawlak, J. K. Abramski, J. Ferenc, L. S. Foteeva, A. R. Timerbaev, B. K. Keppler and M. Jarosz, J. Chromatogr., 2008, 1192, 323–326 CrossRef CAS.
- A. R. Timerbaev and B. K. Keppler, Anal. Biochem., 2007, 369, 1–7 CrossRef CAS.
- M. Groessl, C. G. Hartinger, P. J. Dyson and B. K. Keppler, J. Inorg. Biochem., 2008, 102, 1060–1065 CrossRef CAS.
- M. Sulyok, S. Hann, C. G. Hartinger, B. K. Keppler, G. Stingeder and G. Koellensperger, J. Anal. At. Spectrom., 2005, 20, 856–863 RSC.
- A. Levina, R. S. Armstrong and P. A. Lay, Coord. Chem. Rev., 2005, 249, 141–160 CrossRef CAS.
- I. Ascone, R. Fourme, S. Hasnain and K. Hodgson, J. Synchrotron Radiat., 2005, 12, 1–3 CAS.
- J. E. Penner-Hahn, Coord. Chem. Rev., 2005, 249, 161–177 CrossRef CAS.
- G. N. George, I. J. Pickering, C. J. Doonan, M. Korbas, S. P. Singh and R. E. Hoffmeyer, Adv. Mol. Toxicol., 2008, 2, 123–152 Search PubMed.
- W. Shi and M. R. Chance, Cell. Mol. Life Sci., 2008, 65, 3040–3048 CrossRef CAS.
- M. D. Hall, C. T. Dillon, M. Zhang, P. Beale, Z. Cai, B. Lai, A. P. J. Stampfl and T. W. Hambley, JBIC, J. Biol. Inorg. Chem., 2003, 8, 726–732 CrossRef CAS.
- M. D. Hall, G. J. Foran, M. Zhang, P. J. Beale and T. W. Hambley, J. Am. Chem. Soc., 2003, 125, 7524–7525 CrossRef CAS.
- I. Ascone, L. Messori, A. Casini, C. Gabbiani, A. Balerna, F. Dell'Unto and A. Congiu Castellano, Inorg. Chem., 2008, 47, 8629–8634 CrossRef CAS.
- T. W. Kallen and J. E. Earley, Inorg. Chem., 1971, 10, 1149–1151 CrossRef.
- H. Taube, Comments Inorg. Chem., 1981, 1, 17–31 CAS.
- A. Levina, H. H. Harris and P. A. Lay, J. Am. Chem. Soc., 2007, 129, 1065–1075 CrossRef CAS; A. Levina, H. H. Harris and P. A. Lay, J. Am. Chem. Soc., 2007, 129, 9832 CrossRef CAS.
- A. Nguyen, I. Mulyani, A. Levina and P. A. Lay, Inorg. Chem., 2008, 47, 4299–4309 CrossRef CAS.
- A. Levina and P. A. Lay, Chem. Res. Toxicol., 2008, 21, 563–571 CrossRef CAS.
- T. Paunesku, S. Vogt, J. Maser, B. Lai and G. Woloschak, J. Cell. Biochem., 2006, 99, 1489–1502 CrossRef CAS.
- R. A. Alderden, H. R. Mellor, S. Modok, M. D. Hall, S. R. Sutton, M. G. Newville, R. Callaghan and T. W. Hambley, J. Am. Chem. Soc., 2007, 129, 13400–13401 CrossRef CAS.
- F. M. Verbi, S. C. C. Arruda, A. P. M. Rodriguez, C. A. Pérez and M. A. Z. Arruda, J. Biochem. Biophys. Methods, 2005, 62, 97–109 CrossRef CAS.
- H. H. Harris, A. Levina, C. T. Dillon, I. Mulyani, B. Lai, Z. Cai and P. A. Lay, JBIC, J. Biol. Inorg. Chem., 2005, 10, 105–118 CrossRef CAS.
- K. L. Munro, A. Mariana, A. I. Klavins, A. J. Foster, B. Lai, S. Vogt, Z. Cai, H. H. Harris and C. T. Dillon, Chem. Res. Toxicol., 2008, 21, 1760–1769 CrossRef CAS.
- J. B. Aitken, E. A. Carter, H. Eastgate, M. J. Hackett, H. H. Harris, A. Levina, Y.-C. Lee, C.-I. Chen, B. Lai, S. Vogt and P. A. Lay, Radiat. Phys. Chem., 2009 DOI:10.1016/j.radphyschem.2009.03.068.
- E. A. Carter, K. K. Tam, R. S. Armstrong and P. A. Lay, Biophys. Rev., 2009, 1, 95–103 Search PubMed.
| This journal is © The Royal Society of Chemistry 2009 |