Interplay of metal ions and urease
Eric L.
Carter
a,
Nicholas
Flugga
a,
Jodi L.
Boer
b,
Scott B.
Mulrooney
a and
Robert P.
Hausinger
*abc
aDepartment of Microbiology and Molecular Genetics, Michigan State University, East Lansing, Michigan 48824-4320, USA. E-mail: hausinge@msu.edu
bDepartment of Biochemistry and Molecular Biology, Michigan State University, East Lansing, Michigan 48824-1319, USA
cQuantitative Biology Program, Michigan State University, East Lansing, Michigan 48824-4320, USA
First published on 9th April 2009
Abstract
Urease, the first enzyme to be crystallized, contains a dinuclear nickel metallocenter that catalyzes the decomposition of urea to produce ammonia, a reaction of great agricultural and medical importance. Several mechanisms of urease catalysis have been proposed on the basis of enzyme crystal structures, model complexes, and computational efforts, but the precise steps in catalysis and the requirement of nickel versus other metals remain unclear. Purified bacterial urease is partially activated via incubation with carbon dioxide plus nickel ions; however, in vitro activation also has been achieved with manganese and cobalt. In vivo activation of most ureases requires accessory proteins that function as nickel metallochaperones and GTP-dependent molecular chaperones or play other roles in the maturation process. In addition, some microorganisms control their levels of urease by metal ion-dependent regulatory mechanisms.
 Eric L. Carter Eric L. Carter | Eric L. Carter is a graduate student who received his BS in Biomedical Science from Western Michigan University in 2005 then worked as a research technician at Midwestern University until 2007. His area of research focuses on the involvement of UreD in urease activation and testing of a putative nickel-independent urease. |
 Nicholas Flugga Nicholas Flugga | Nicholas Flugga is a graduate student who received his BS in Biology at the University of Findlay in 2007. His research focuses on testing whether UreB shifts by a hinge-like motion to allow access to the nascent active site during urease activation. |
 Jodi L. Boer Jodi L. Boer | Jodi L. Boer is a graduate student who received her BS at Calvin College in 2007. She carries out structural and biochemical studies of UreG, required for activation of this nickel-containing enzyme. |
 Scott B. Mulrooney Scott B. Mulrooney | Scott B. Mulrooney received his PhD from MSU in 1990, was a postdoctoral fellow and lecturer at the University of Michigan, and returned to MSU in 1999 where he is now an Associate Research Professor. His urease studies have focused on characterization of UreE. |
 Robert P. Hausinger Robert P. Hausinger | Robert P. Hausinger received his PhD at the University of Minnesota in 1982. After postdoctoral training at M.I.T. (1982–1984) he joined the faculty at Michigan State University where he is Professor of Microbiology and Biochemistry, and Director of the Quantitative Biology Program. His research focuses on the mechanisms of metallocenter biosynthesis and the catalytic mechanisms and versatility of FeII/α-ketoglutarate hydroxylases. |
1.0 Introduction to urease
Urease holds a prominent place in the history of Science. It was the first enzyme to be crystallized1 and the first shown to contain nickel.2 The enzyme catalyzes the deceptively simple hydrolysis of urea into ammonia and carbamic acid. The latter compound spontaneously decomposes into carbonic acid and a second ammonia molecule, as shown in the reactions below:| H2N–C(O)–NH2 + H2O → NH3 + H2N–COOH |
| H2N–COOH + H2O → NH3 + H2CO3 |
Ureases have been isolated and characterized from various bacteria, fungi, and plants.3,4,11–13 Despite having differences in their quaternary structures, ureases possess essentially identical folds and are united by a basic trimeric structure containing three catalytic centers.14 Most bacterial ureases, exemplified by those from Klebsiella aerogenes (Fig. 1A)15 and Bacillus pasteurii,16 possess three distinct types of subunits that form a (UreABC)3 structure. Urease from H. pylori has only two subunits, the smaller of which is equivalent to a fusion of UreA and UreB found in other bacterial enzymes . The two H. pyloriurease subunits form a trimer of dimers that closely resembles other ureases, but this unit forms a supramolecular ((UreAB)3)4 complex as shown in Fig. 1B.17 Eukaryotic organisms exhibit yet another configuration, as illustrated by jack bean (Canavalia ensiformis) urease—the enzyme used in the pioneering studies mentioned above. This plant enzyme contains a single type of subunit which represents a fusion of the separate subunits found in bacterial ureases. The subunit assembles into a trimer analogous to the (UreABC)3 trimer shown for the K. aerogenesenzyme (Fig. 1A), and two of these trimers dimerize in a face-to-face manner to generate the native hexameric structure (rotated 90 degrees relative to the other enzymes in Fig. 1C).18
![The structures of three well-characterized ureases. (A) K. aerogenesurease (PDB access code 1fwj) with UreA depicted in blue, UreB in orange, and UreC in yellow, together forming a (UreABC)3 structure. (B) H. pyloriurease (1e9z) with UreA (corresponding to a fusion of the two small subunits in the K. aerogenesenzyme) depicted in blue and UreB (analogous to UreC in the K. aerogenesprotein) shown in yellow for one (UreAB)3 unit, with three more (UreAB)3 units shown in gray included in the biologically relevant [(UreAB)3]4 structure. (C) Jack bean urease with one subunit (comparable to a fusion of all three K. aerogenes subunits) shown in gold in the otherwise blue hexameric protein (two trimers interacting in a face-to-face manner and shown after a 90 degree rotation compared to the other ureases). (Copied by permission of the International Union of Crystallography; http://journals.iucr.org/).](/image/article/2009/MT/b903311d/b903311d-f1.gif) | ||
| Fig. 1 The structures of three well-characterized ureases. (A) K. aerogenesurease (PDB access code 1fwj) with UreA depicted in blue, UreB in orange, and UreC in yellow, together forming a (UreABC)3 structure. (B) H. pyloriurease (1e9z) with UreA (corresponding to a fusion of the two small subunits in the K. aerogenesenzyme ) depicted in blue and UreB (analogous to UreC in the K. aerogenesprotein) shown in yellow for one (UreAB)3 unit, with three more (UreAB)3 units shown in gray included in the biologically relevant [(UreAB)3]4 structure. (C) Jack bean urease with one subunit (comparable to a fusion of all three K. aerogenes subunits) shown in gold in the otherwise blue hexameric protein (two trimers interacting in a face-to-face manner and shown after a 90 degree rotation compared to the other ureases). (Copied by permission of the International Union of Crystallography; http://journals.iucr.org/). | ||
The active site of urease contains two nickel atoms bridged by a carboxylated lysine and a solvent molecule, as shown for the K. aerogenesenzyme (Fig. 2).19 In addition, Ni1 is coordinated by two His residues plus a terminal water molecule while Ni2 is coordinated by two His, an Asp, and a terminal solvent. Also present at the active site are two His residues thought to participate in substrate binding and/or catalysis. This active site structure is generally consistent with results derived from earlier biophysical investigations of the metallocenter. X-Ray absorption spectroscopy (XAS) of jack bean,20,21K. aerogenes,22 and B. pasteurii23ureases provides evidence for 5–6 coordinate nickel ions with exclusively O/N ligands, including about two imidazoles per metal, and the K. aerogenes data include a scattering component indicating a Ni–Ni distance of ∼3.26 Å.22 The presence of β-mercaptoethanol leads to thiolate-to-nickel charge-transfer transitions,24,25 a Ni–S scattering interaction seen by XAS,21,22 and a unique variable-temperature magnetic circular dichroism feature26 that are all consistent with the inhibitor sulfur atom replacing the bridging solvent molecule in the dinuclear metallocenter. Structural characterization of the β-mercaptoethanol-bound enzyme directly demonstrates this binding mode.27 Preliminary studies to characterize the magnetic properties of the metals in jack bean urease suggest weak antiferromagnetic exchange coupling between the nickel atoms,28 but saturation magnetization data collected with both jack bean and K. aerogenesureases fail to confirm this result.
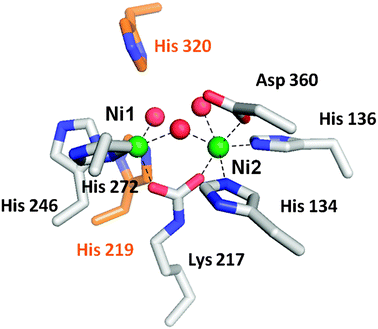 | ||
| Fig. 2 The urease active site. The active site of urease contains two nickel atoms (green) bridged by a carboxylated lysine and a hydroxyl group. Ni1 is also coordinated by two histidine residues and a solvent molecule, while Ni2 is coordinated by two histidines, an aspartic acid residue, and a water molecule. Waters are red, metal-binding side chains are shown with white carbon atoms, and two nearby histidine residues that function in catalysis are shown with orange carbon atoms. | ||
Many residues nearby or directly coordinating to the K. aerogenesurease dinuclear active site have been substituted by mutagenesis approaches, and structures are known for several of these variants.19,29–31 The H134A variant is notable because removal of the Ni2 ligand results in formation of a mononuclear Ni1 site.29 Also of interest are studies where the lysine residue that becomes carboxylated in the wild-type enzyme is substituted. The K217A and K217C variants are inactive as isolated, but activity is generated by incubating the proteins with nickel ions plus formic acid. Structures of the chemically rescued enzymes are known.19
In this review, we discuss the function of nickel in urease catalysis, describe how ureaseapoprotein can be reconstituted in vitro with nickel and other metal ions, summarize our current understanding of how urease is activated in vivo in bacteria, fungi, and plants, and highlight ureasegenes that exhibit metal-dependent regulation.
2.0 Role of nickel in urease
Soon after the discovery that urease contains two nickel ions per active site, Dixon et al. proposed a catalytic mechanism incorporating both metals.32 These researchers hypothesized that one metal site binds the urea carbonyl oxygen and enhances the electrophilicity of the carbon atom, the second metal increases the nucleophilicity of a water molecule, and protein side chains serve additional acid/base roles to achieve urea hydrolysis. Notably, this hydrolysis reaction is distinct from the non-catalyzed decomposition of urea which proceeds by elimination of ammonia with consequent formation of cyanic acid (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CN–H), a compound that is not observed in enzymatic reactions. Informed by the structure of the K. aerogenesenzyme ,15 the initial hydrolytic mechanism has been elaborated as shown in Fig. 3A. In this model, the urea carbonyl oxygen is suggested to displace a terminal water molecule from the five-coordinate nickel (Ni1), with additional stabilization offered by interaction with His 219 (K. aerogenes numbering). The terminal solvent bound to the hexacoordinate nickel (Ni2) is thought to be appropriately positioned to attack the carbonyl oxygen, forming a tetrahedral intermediate that decomposes with His 320 acting as a general acid. Support for the binding of the urea carbonyl oxygen to Ni1 is derived from the structure of acetohydroxamic acid-bound enzyme , where this type of interaction is observed.19 This simple scheme nicely accounts for the catalytic properties of H219A and H320A variants of the protein,31 and this mechanism accommodates the bell-shaped pH dependence of the protein if one invokes a reverse-protonation model in which activity requires the protonated state of His 320 (pKa ∼ 6.5) and the deprotonated state of the solvent bound to Ni2 (pKa ∼ 9).33
CN–H), a compound that is not observed in enzymatic reactions. Informed by the structure of the K. aerogenesenzyme ,15 the initial hydrolytic mechanism has been elaborated as shown in Fig. 3A. In this model, the urea carbonyl oxygen is suggested to displace a terminal water molecule from the five-coordinate nickel (Ni1), with additional stabilization offered by interaction with His 219 (K. aerogenes numbering). The terminal solvent bound to the hexacoordinate nickel (Ni2) is thought to be appropriately positioned to attack the carbonyl oxygen, forming a tetrahedral intermediate that decomposes with His 320 acting as a general acid. Support for the binding of the urea carbonyl oxygen to Ni1 is derived from the structure of acetohydroxamic acid-bound enzyme , where this type of interaction is observed.19 This simple scheme nicely accounts for the catalytic properties of H219A and H320A variants of the protein,31 and this mechanism accommodates the bell-shaped pH dependence of the protein if one invokes a reverse-protonation model in which activity requires the protonated state of His 320 (pKa ∼ 6.5) and the deprotonated state of the solvent bound to Ni2 (pKa ∼ 9).33
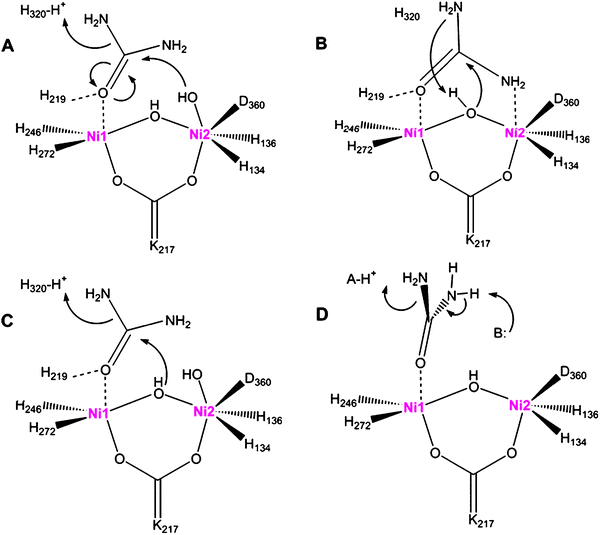 | ||
| Fig. 3 Proposals for the urease catalytic mechanism. (A) The hydroxyl group bound to Ni2 attacks urea, whose carbonyl group is polarized by coordination to Ni1, forming a tetrahedral intermediate that releases ammonia with His 320 (K. aerogenes numbering) acting as a general acid. (B) The bridging hydroxyl group attacks urea, bound with its carbonyl group coordinated to Ni1 and an amine interacting with Ni2, and the hydroxyl proton transfers to the released ammonia. (C) A merged mechanism in which the bridging water attacks the substrate, but with His 320 acting as a general acid. (D) Elimination mechanism to form a cyanic acid (OCN–H) intermediate that subsequently becomes hydrated (not depicted) to form carbamate. In all mechanisms, the carbamate spontaneously decomposes. | ||
An alternative mechanism for urea hydrolysis was proposed on the basis of a structure determined for phenylphosphorodiamidate-inhibited urease of B. pasteurii.16 The inhibitor is in fact a slow substrate of the enzyme ,34 with release of phenol leading to the diamidophosphoric acid-bound form of the enzyme . An amino group of diamidophosphoric acid is bound to Ni2, leading the investigators to suggest that binding of urea displaces both terminal water molecules from the dinuclear center to coordinate in a bidentate fashion (Fig. 3B). Significantly, the structure is consistent with attack of the initial inhibitor by the bridging (not terminal) water molecule. The bridging solvent molecule is thus proposed to attack the urea carbonyl carbon during normal catalysis.
A merged mechanism has also been considered (Fig. 3C). In this case, the bridging water attacks the substrate, but with His 320 continuing to function as a general acid. This model readily accommodates the results of mutagenesis studies,31 whereas large changes in the kinetics of His 320 variants fit less clearly to the model of Fig. 3B. Fluoride inhibition studies of K. aerogenesurease were interpreted in terms of the halogen replacing the bridging water molecule to inhibit the enzyme , thus providing support to the hypothesis that the bridging water serves as the nucleophile .35
Examination of biomimetic model compounds has provided further insight into the chemistry available to urease-like dinuclear nickel sites. A wide assortment of such studies has been reported, only three of which are briefly highlighted here. In the first example, the ligand N,N,N′,N′-tetrakis[(6-methyl-2-pyridyl)methyl]-1,3-diaminopropan-2-ol (Me4-Htpdp) was used to prepare [Ni2(Me4-tpdp)(CH3CO2)(ClO4)(CH3OH)]ClO4 whose structure is known.36Urea addition leads to displacement of ClO4− as the urea carbonyl oxygen coordinates to one nickel, verified by crystallography, with accompanying changes in the absorption spectrum. Of functional significance, this species reacts with ethanol to generate ethyl carbamate. For the second representative example, a pyrazolate-based ligand (HL2) was used to synthesize the crystallographically characterized L2Ni2(O2H3)(ClO4)2 species.37 The addition of urea leads to the displacement of H3O2 and bidentate binding with the carbonyl oxygen coordinating to one nickel and an amine binding to the second. Heating of this sample leads to the elimination of ammonia and provides a bridging cyanato complex. Finally, 1,4-bis(2,2′-dipyridylmethyl)phthalazine (bdptz) was used to synthesize [Ni2(μ-urea)(bdptz)(urea)(CH3CN)](ClO4)3 as a dinuclear urease mimic.38 Heating of this compound results in the elimination of ammonia, followed by slow hydration of the resulting cyanate, leading the investigators to suggest that such a sequence of reactions could also account for the ammonia and carbamate products of urease (Fig. 3D).
To further define the mechanism of urease, high level computational methods have been investigated. For example, shortly after publication of the structure of diamidophosphoric acid-inhibited B. pasteuriiurease, ab initio and density functional theory approaches were used to explore the most likely pathway of urea hydrolysis.39 These studies suggest that the urea carbonyl oxygen first coordinates to Ni1, with a urea amine binding to Ni2 concomitantly with, or subsequent to, attack of the bridging hydroxide on the urea carbonyl. Later quantum chemical calculations of dinuclear nickel complexes are consistent with the bridging ligand being a hydroxide rather than an oxo dianion and suggest that urea can bind in a monodentate or bidentate fashion.40 Computational analysis of the bdptz complex mentioned above led to the suggestion that urea may decompose by both hydrolytic and elimination routes.41 Molecular dynamics simulations were used to both assess the likely protonation status of His 219 and His 320 and compare various urea decomposition pathways.42 More recent quantum chemical studies extend the examination of various pathways and come to the same conclusion as above that hydrolysis and elimination reactions may compete in the enzyme .43 Several alternative elimination pathways were proposed with the various mechanisms utilizing different general base and general acid groups (Fig. 3D), but all forming cyanate as a product. Significantly, however, cyanate has not been detected during the urease reaction nor is it a substrate of the enzyme (unpublished observations). It is feasible that cyanate is not released from the enzyme , but rather that the metallocenter-bound cyanate is immediately hydrated, analogous to the situation reported for the bdptz model complex,38 but calculations related to this additional step have not been reported.
3.0 In vitro reconstitution and properties of metal-substituted ureases
Of far-reaching significance, the incubation of purified K. aerogenesureaseapoprotein with nickel ions and carbon dioxide results in partially activated enzyme .44 The properties of this in vitro activation process are extensively characterized and have set the stage for comparison to similar studies with various ureaseapoprotein-containing complexes (section 4.0) and for understanding the in vivo mechanism of enzyme maturation. Simple addition of nickel ions to ureaseapoprotein does not generate detectable activity, whereas the use of buffers containing bicarbonate (which is in equilibrium with CO2 in solution) plus nickel ions yields active urease in approximately 15% of the protein.44,45CO2 rather than bicarbonate is the critical reagent required for this process as shown by using stock bicarbonate buffers at pH 4 or 8.5 during short-term activation experiments (with the same final pH 8.3 conditions); the low pH stock solution provides more rapid production of active urease because the more acidic conditions result in higher CO2 concentrations.44 Inclusion of carbonic anhydrase when using the low-pH-bicarbonate stock solution decreases the activation rates, further supporting the involvement of CO2 in the activation process. Using buffers of varied pH, all supplied with 0.3% CO2, the extent of activation increases with pH consistent with a process involving a group with a pKa of at least 9. These findings led to the suggestion that the deprotonated form of an active site residue of high pKa reacts with CO2 to provide a metal-binding ligand needed to form active enzyme . Elucidation of the urease crystal structure revealed the carboxylated Lys bridging ligand (Fig. 2), in perfect agreement with these results.Additional studies have characterized the kinetics of the activation process and the timing of nickel addition. After identifying optimal concentrations of bicarbonate (100 mM) and nickel ions (100 μM) for activation at pH 8.3, the process still requires about 90 min to reach maximum activity.44 These results demonstrate the relative inaccessibility of the nascent active site to bicarbonate and metal ions, and they highlight the importance of additional components in generating urease activity during cell growth. Use of radiolabeled bicarbonate demonstrates that about 50% of the protein is stably carboxylated after activation; notably, this value is significantly larger than the percent of enzyme that is active. When ureaseapoprotein is first incubated with nickel ions and then exposed to bicarbonate, no activation is observed.45 This finding indicates that CO2 binding must precede binding of Ni. Metal analysis indicates that this nickel-inhibited species and the protein subjected to optimal activation conditions both contain nearly two bound nickel ions per active site even though only 15% of the latter species is active. The improperly bound nickel ions are more labile than the active metallocenters, which are stable to prolonged incubation with EDTA. Extended exposure of the activated protein to EDTA removes ∼75% of the bound nickel without affecting activity, and a second round of activation provides a further increase in the levels of activity.45,46XAS studies show the metallocenter properties of the inactive nickel-containing urease and the active enzyme are nearly indistinguishable.46 The metallocenter structure of the nickel- and bicarbonate-treated, but still inactive, protein species remains unclear.
The interaction of ureaseapoprotein with other metal ions also has been examined. Pre-treatment of ureaseapoprotein with zinc, copper, cobalt, or manganese leads to inhibition of the normal CO2 and nickel-dependent activation, whereas magnesium and calcium have no effect on this process.45 In the absence of nickel, incubation of apoprotein with manganese ions plus bicarbonate leads to activity levels comparable to activation of about 2% of the urease active sites.45 In contrast to the situation with nickel, the active mangano-urease loses activity when incubated with EDTA. The EDTA-treated protein still contains ∼0.4 manganese per active site and this inactive protein was able to be crystallized.46 The structure of this dinuclear manganese metallocenter is nearly indistinguishable from that of the active nickel-containing enzyme and the reason for its inactivity is unknown. In contrast to the situation with manganese, incubation of apoprotein with cobalt plus bicarbonate yields no activity; however, the electronic spectrum of this species possesses a feature that could arise from a thiolate-to-cobalt charge-transfer transition.46 When the same study is carried out using a variant apoprotein (C319A) lacking a cysteine residue near the active site, no charge-transfer band is observed and significant levels of activity (∼2% of similarly treated wild-type apoprotein) are measured. XAS studies are consistent with partial thiolate coordination in the sample generated from wild-type protein, and this ligand is missing in the C319A mutant. The manganese and cobalt results demonstrate that ureaseapoprotein can acquire some activity with non-nickel metal ions when they are appropriately incorporated into the protein. In contrast, zinc- and copper-bound urease are inactive under all conditions tested.45,46
SO2, CS2, and vanadate were investigated for their capacities to substitute for CO2 in serving as lysine modification reagents.47SO2 neither inhibits the normal activation process nor substitutes for CO2. By contrast, CS2 inhibits standard activation and when incubated with nickel ions plus ureaseapoprotein it generates a species containing a sulfur-to-nickel charge-transfer transition. These results are interpreted in terms of formation of a lysine dithiocarbamate-bridged metallocenter. Remarkably, when ureaseapoprotein is incubated with nickel ions plus vanadate, activity is generated at levels similar to those obtained with bicarbonate.47 These results suggest the formation of a vanadylated lysine bridging two nickel ions at the active site.
In contrast to the detailed in vitro activation studies with K. aerogenesureaseapoprotein, very limited findings are available with other ureases. As one example, nickel-containing jack bean urease was dialyzed at room temperature against buffers containing zinc and cobalt for several weeks.48 After 51 days of dialysis versuszinc ions the nickel content of the protein reduces from 2 to 0.89 per active site, the zinc content increases to 0.83 per active site, and the activity drops to 10% of the starting value. Similarly, after 87 days of dialysis versus cobalt ions the nickel content drops to 1.27 per active site, the cobalt content increases to 0.75 per active site, and the activity diminishes to 27%. These results were interpreted in terms of the production of mixed-metal derivatives, but additional metallocenter studies (such as XAS) are needed to confirm this conclusion. More generally, the very lengthy time periods involved in these studies raise questions about the physiological relevance of these metal substitution studies.
4.0 In vivo maturation of bacterial ureases
Only a portion of purified ureaseapoprotein acquires activity in vitro (∼15% in the case of the K. aerogenesprotein; section 3.0), even when using optimized activation conditions. In the bacterial cell, this limitation is overcome by the concerted effort of a series of accessory proteins.49,50 In this section, we describe how auxiliary genes encoding accessory proteins are often clustered with the structural genes for urease, we detail the properties of each gene product and its associated protein complexes, and we briefly mention additional genes that affect urease activation in some microorganisms.4.1 Genetic structure of urease-associated genes
Bacterial urease structural genes often are clustered with genes encoding urease-associated proteins, but their number and order are not universal across species (Fig. 4). For example, the three genes encoding urease (ureABC) of K. aerogenes are flanked by four genes (ureD positioned upstream and ureE, ureF, and ureG found downstream) encoding accessory proteins, each of which facilitates urease activation.51,52 This ureasegene structure is retained in Rhizobium leguminosarum, but with several uncharacterized open reading frames (ORFs) inserted into this cluster.53 Many bacteria position ureD after ureG, as shown for thermophilic Bacillus sp. TB-90 which also contains the downstream ureH (likely encoding a nickel ion permease) and another ORF.54 In Actinobacillus pleuropneumoniae, the ureABCEFGD cluster is interrupted by an ORF and is preceded by a gene cluster encoding a five-component nickel transporter and a possible urea permease.55 The H. pyloriurease cluster consists of ureA, a fusion of the small subunit genes from other bacteria, and ureB, encoding the large subunit,56,57 along with five downstream genes: ureI (encoding a proton-gated urea channel),58ureE, ureF, ureG, and ureH (homologous to ureD of other bacteria).59 Some microorganisms, such as Helicobacter mustelae,60 contain a complete ureasegene cluster as well as a second set of structural genes not associated with any accessory proteingenes. Furthermore, some bacteria lack one or more of the four typical urease auxiliary genes; this is best exemplified by Bacillus subtilis which lacks any identifiable accessory proteingenes.61 The latter result suggests that accessory proteins are not always required for in vivourease activation or that genetically unlinked cellular maturation factors are utilized in some cases. Below, we summarize evidence demonstrating the need for four auxiliary genes in most ureolytic bacteria, we describe the properties and functions of UreD, UreF, UreG, and UreE using the framework illustrated in Fig. 5, and we discuss evidence for additional genes needed for urease production in selected systems. Unless clearly indicated otherwise, the results described in the following sections involve analysis of K. aerogenesureasegenes expressed in E. coli.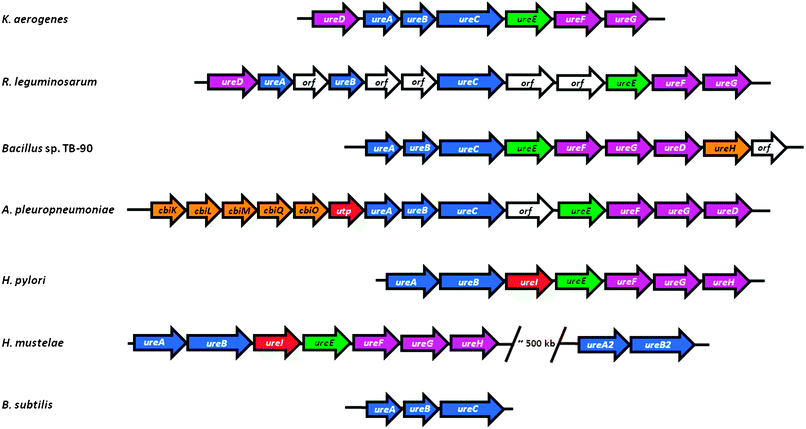 | ||
| Fig. 4 Organization of representative ureasegene clusters. The ureasegene cluster of K. aerogenes (ureDABCEFG) is compared to the gene organization found in selected other bacteria: R. leguminosarum, containing insertions of unidentified genes; Bacillus sp. TB-90, which repositions ureD and adds a likely nickel permease (ureH); A. pleuropneumoniae, which juxtaposes genes encoding a likely nickel transport (cbiKLMO) and urea permease (utp); Helicobacter species which fuse the two small structural subunits and add a proton-gated urea channel (ureI); H. mustelae containing a second, incomplete urease cluster; and B. subtilis that lacks urease accessory genes. Genes encoding urease subunits are shown in blue, the ureEgene encoding a metallochaperone is green, other urease accessory genes are purple, genes encoding proteins involved in nickel uptake are orange, those encoding proteins related to urea transport are red, and unknown genes are white. The sizes of the arrows do not accurately reflect the sizes of the genes. | ||
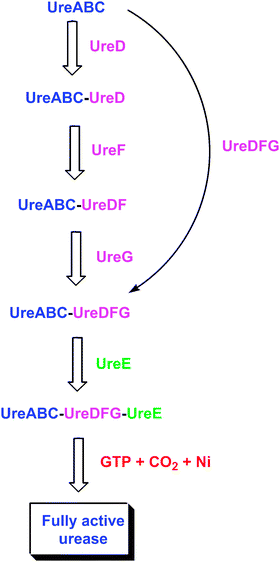 | ||
| Fig. 5 Postulated interactions among urease-related bacterial proteins. The ureaseapoprotein sequentially binds UreD, UreF, and UreG, and the in vitro activation properties exhibit differences in each apoprotein species. Alternatively, a preformed UreDFG complex may bind the apoprotein. Within the resulting complex, the UreDFG heterotrimer acts as GTP-dependent molecular chaperone to enhance exposure of the nascent active site. UreE interacts with the UreABC-UreDFG complex and delivers nickel ions, thus serving a metallochaperone role. Carbon dioxide is used to form the carboxy-lysine metal ligand, and GTP hydrolysis (occurring in UreG) drives the metallocenter assembly process to provide active urease, with release of all accessory proteins. | ||
4.2 In vivo demonstration that accessory proteins are required
The significance of the individual accessory proteins in urease activation has been assessed in the K. aerogenes system by using a systematic knockout and complementation approach. Each auxiliary gene was at least partially deleted in the context of the otherwise intact ureDABCEFGgene cluster, and the urease activities in cell extracts were compared to those in cells expressing the wild-type genes [approximately 200 μmol urea degraded min−1 (mg protein)−1 or ∼200 U mg−1 in cell extracts].51,62 It was shown that ureD, ureF, and ureG are all essential for the production of functional urease since mutations in these genes nearly abolish activity in cell extracts (<1 U mg−1).51 In these early studies, partial deletions in ureE resulted in only ∼50% lower specific activities; however, subsequent efforts with a true ureE deletion mutant show that urease activity is essentially eliminated.63 Furthermore, E. coli cells expressing only the K. aerogenes structural genes and grown in the presence of 5 mM NiCl2 possess only trace levels (<0.05 U mg−1) of urease activity.61 Significantly, purified urease from cells expressing the deletion mutants of ureD, ureF, and ureG contain negligible amounts of nickel. Urease activity is largely restored to cells co-expressing the mutant gene clusters from one plasmid with a wild-type complement of the mutant gene on a second plasmid.51 Taken together these data indicate that each of the four K. aerogenes accessory proteins plays an essential role in urease maturation.Genetic experiments using deletion, insertional inactivation, and complementation approaches have been used to identify multiple non-urease-subunit genes required for urease activity in Proteus mirabilis,64Klebsiella pneumoniae,65Providencia stuartii,66 and many other bacteria, including the peptic-ulcer causing H. pylori. In the latter case, disruptions or deletions in ureA, ureB, ureF, ureG, or ureH of the H. pyloriureasegenes expressed in E. coli result in a non-ureolytic phenotype .59 Subsequent studies confirm these results and further show a significant reduction in urease activity in cells lacking ureE.67,68 Detailed investigations of the four standard accessory proteins are discussed in sections 4.3–4.6, and the products of additional genes found in selected microorganisms are summarized in section 4.7.
4.3 UreD and the UreABC–UreD complex
Attempts to heterologously express K. aerogenes ureD in E. coli lead to the production of inclusion bodies,69 and the resulting absence of a soluble protein prevents characterization of the general protein properties. Recently, however, a maltose binding protein (MBP)-UreD fusion protein was found to be soluble and its characterization is in progress (Carter and Hausinger, unpublished observations). No purification of UreD from another bacterium is reported, and no structure of UreD is available. In contrast to the dearth of information about isolated UreD, the properties of several protein complexes containing this accessory protein (Fig. 5) are known, as discussed below.When ureD is highly expressed along with the genes encoding the urease subunits, a UreABC–UreD protein complex can be purified.69 This complex is known by comparison of native and denaturing gel electrophoresis to be comprised of four species, where the (UreABC)3apoprotein binds to 0–3 molecules of UreD. Results from chemical cross-linking studies indicate that UreD is in close proximity to both UreB and UreC,70 and small angle X-ray absorption (SAXS) studies confirm the binding of UreD near UreB in the UreABC–UreD complex.71 When the K. aerogenes UreABC–UreD complex is subjected to the in vitro activation conditions described in section 3, about 30% of the nascent active sites are activated,49,69 demonstrating that UreD significantly enhances the activation competence over that of the apo-protein alone. Following activation, UreD dissociates from the structural subunits. Taken together these data indicate that UreD plays an important, but still undefined, role in incorporating nickel into the urease active site.
Corroborating the results observed in the K. aerogenes system, yeast two-hybrid studies of H. pyloriproteins identify interactions between the corresponding UreH and UreA proteins.67,72 Furthermore, immunoprecipitation of UreC from E. coli cell extracts expressing the P. mirabilisureasegenes leads to coprecipitation of UreD, consistent with an interaction between these two proteins.73
4.4 UreF and the UreABC–UreDF complex
Similar to the situation for ureD, heterologous expression of K. aerogenes ureF leads to an insoluble protein.74 In this case, however, two soluble forms of UreF have been described: an MBP–UreF fusion75 and a UreE–UreF fusion.76 The general properties of the MBP–UreF fusion protein and its capacity to interact with other ureaseproteins are unknown. In contrast, the monomeric UreE–UreF fusion protein is capable of facilitating urease activation in the cell, it binds to the UreABC–UreD complex in vitro, and it forms an even larger complex in vivo.76 No UreF protein has been purified from any other microorganism, and no structure is reported. Nevertheless, a homology model was created for a portion of UreF from B. pasteurii by using two GTPase activating enzymes as templates, and a similar role is hypothesized for this protein.77 Like UreD, UreF is a component of a number of urease-related protein complexes (Fig. 5), as described below.Over-expression of ureF and ureD along with ureABC leads to the production of a UreABC–UreDF complex.74 In contrast, no complex is generated by expressing just ureF with ureABC. The purified UreABC–UreDF complex is a mixture of species containing 0–3 UreDF heterodimers per (UreABC)3apoprotein according to Western blot analysis of native gels using anti-urease antibodies.74 Significantly, anti-UreD antibodies fail to detect UreABC–UreDF complexes, but do recognize UreABC–UreD, thus suggesting that UreF masks the UreD epitopes by directly binding to UreD. Support for this proposal is derived from a study involving chemical cross-linking in conjunction with matrix-associated laser desorption-ionization mass spectrometry of proteolytic fragments.70 No UreD cross-links are observed in the UreABC–UreDF complex whereas distinct cross-links are detected in the UreABC–UreD species. Further analysis by this cross-linking/proteolysis/mass spectrometry approach provides evidence for an altered conformation of the UreABC–UreDF complex in comparison to UreABC–UreD or ureaseapoprotein. In particular, a shift of UreB in a hinge-like motion is hypothesized in order to account for cross-linking of a lysine residue in UreB with another lysine in a distant portion of UreC (located over 50 Å away in the ureaseapoprotein). Compatible with this proposal, flexibility analysis of urease demonstrates that UreB can reasonably shift to enhance access to the nascent active site.71SAXS studies lack sufficient resolution to address the presence of a shifted UreB domain, but the modeled structure derived from these data places UreD and UreF together in close contact with UreB.71 In aggregate, these results are consistent with UreABC–UreDF being capable of undergoing a conformational change that enhances its competence for insertion of bicarbonate and Ni into the buried active site by increasing its exposure. UreABC–UreDF is similar to the UreABC–UreD complex in allowing ∼30% of the available protein to be activated using standard conditions; however, the UreABC–UreDF complex requires lower concentrations of bicarbonate for activation because it is less susceptible to nickel-dependent inactivation. The protection against nickel inhibition suggests a role for UreF in regulating the sequential incorporation of bicarbonate before nickel into the active site.
Although UreABC–UreDF complexes have not been studied from other microorganisms, UreD–UreF interactions have been noted. For example, yeast two-hybrid analysis reveal the association of UreD with UreF in P. mirabilis.73 This same approach demonstrates an interaction between UreF and UreH, an orthologue of UreD, in the H. pylori system.67,72
4.5 UreG and its complexes
UreG is a soluble protein that has been purified and characterized from several microorganisms including K. aerogenes,78B. pasteurii,79M. tuberculosis,80 and H. pylori.81 In contrast to the monomeric protein from K. aerogenes, UreG proteins from B. pasteurii and M. tuberculosis are dimeric with the two monomers said to be joined through a disulfide bridge, and that from H. pylori undergoes zinc-dependent (but not nickel-dependent) dimerization. The metal-binding properties of UreG proteins from B. pasteurii and H. pylori are characterized; the former binds two zinc and four nickel ions per dimer79 and the latter binds 0.5 zinc and two nickel ions per monomer.81 Two residues suspected to participate in binding zinc (Cys 66 and His 68) were mutated individually and together; the variant H. pylori UreG proteins bind zinc with a 10-fold larger Kd than wild-type protein and continues to dimerize, but curiously the double mutant protein binds two zinc per dimer.81 The ureG sequences from these and other bacteria reveal a nucleotide binding motif (P-loop) consistent with a GTPase role, but only very low or no hydrolysis of GTP is detected. Mutation of a single residue in this motif for either the K. aerogenes or H. pyloriprotein leads to the abolishment of urease activity.78,82 The B. pasteuriiprotein is claimed to be intrinsically disordered on the basis of NMR dispersion and fluorescence analysis;79,83 however, circular dichroism studies show substantial secondary structure in some members of this protein family. No structure is reported for any UreG, but one has been elucidated for the related protein HypB (required for biosynthesis of nickel-dependent hydrogenases) from Methanocaldococcus jannaschii.84 This dimeric protein contains a GTP binding site and a dinuclear zinc site at the subunit interface. Two of the residues serving as ligands to the HypB zinc site coincide with the UreG residues that had been mutated in the H. pyloriprotein.A UreABC–UreDFG complex (Fig. 5) was identified in E. coli cells expressing a wild-type K. aerogenesurease cluster in the absence of nickel ions.85 UreABC–UreDFG is also obtained in vitro by mixing purified UreG with the isolated UreABC–UreDF complex.86 When activated in the standard buffer containing high levels of nickel and bicarbonate, over 60% of the ureaseapoprotein in the complex is activated, leading to the notion that UreDFG functions as a urease-specific molecular chaperone.87 Importantly, the inclusion of GTP, but not other nucleotide tri-phosphates, results in significant activity when incubated with physiologically relevant bicarbonate levels, compared to the trace activity levels observed in a similar reactions without GTP. Hydrolysis of GTP, and not just binding, is needed for activation as shown by using a non-hydrolyzable GTP analogue. The site of GTP action is within UreG as shown by studies of the UreABC–UreDFG complex containing a mutation in the P-loop motif of UreG; this complex exhibits little competence for activation.86 A hint of a comparable UreG- and urease-containing complex in H. pylori is derived from work in which UreG purified by use of a tandem affinity tag possesses substoichiometric UreB.88
In addition to being a component of the UreABC–UreDFG complex, UreG forms an insoluble, urease-free, UreDFG complex (Fig. 5) in cells expressing ureD, ureF, and ureG and lacking ureE or the structural subunit genes.78 This heterotrimer is solubilized by using low concentrations of detergent (0.5% Triton X-100), allowing for isolation via a combination of anion-exchange chromatography, detergent removal, and use of an ATP-linked agarose column. A UreDFG complex containing a P-loop variant of UreG fails to bind to the nucleotide resin, indicating that an intact P-loop is necessary for this interaction. It is unknown whether pre-formed UreDFG interacts with ureaseapoprotein, and it is unclear whether this complex is physiologically relevant. Nevertheless, the preformed UreDFG species potentially could function as a stable unit during urease activation as an alternative to the sequential binding of UreD, UreF, and UreG to ureaseapoprotein (Fig. 5).
4.6 UreE and the UreABC–UreDFG–UreE complex
The metallochaperone UreE (Fig. 5) delivers nickel to the UreABC–UreDFG complex.87,89,90 This protein has been extensively studied in K. aerogenes where it binds six nickel ions per homo-dimer.91 Most of this nickel is bound to the histidine-rich C-terminus (HGHHHAHHDHHAHSH), but a truncated version (H144*UreE) lacking this region still binds ∼2 Ni2+ atoms per homo-dimer.92 For both proteins, copper, cobalt, and zinc compete with nickel ions, and the spectroscopic properties of these proteins are known.93 For cells grown in the presence of nickel and expressing a urease cluster with H144* UreE substituting for the wild-type version, active urease is produced, indicating that the C-terminus of wild-type UreE is unnecessary for urease activation.91,94 Additional mutagenesis studies suggest that His 110 and His 112 participate in binding nickel but are not critical to urease activation, whereas His 96 has a role in binding the essential metal for transfer to urease.89,94Isothermal titration calorimetry studies of the interaction of nickel, copper, and zinc with H144* UreE demonstrate remarkable complexity, including a protein concentration dependence in these thermodynamic interactions.95 At low protein concentrations, two metal ions bind per dimeric protein; whereas, at high protein concentrations three nickel or copper ions are initially bound per dimer followed by the binding of two additional metal ions. The thermodynamics of nickel ion binding to K. aerogenes H144* UreE is enthalpically favored.The structure of the H144* variant of K. aerogenes UreE dimeric apoprotein is depicted in Fig. 6.96 While the nickel-bound form of the protein is not structurally characterized, the structure of the copper-bound species is known and the protein binds three copper ions. One copper binds at the subunit interface viaHis 96 side chains from each subunit (encircled in red). On the basis of mutational studies, this site is thought to be critical for nickel ion binding related to urease activation.89 Two additional copper ions bind to periphery sites (encircled in blue) viaHis 110 and His 112 side chains. These sites also bind nickel, but mutational studies demonstrate these residues are not essential for urease activation.89 The peripheral sites are suggested to assist in nickel delivery to the central site. Likewise, the His-rich C-terminus of the full-length protein would be positioned well for feeding nickel ions into the interfacial site. The protein domain responsible for binding metal ions resembles a ferredoxin fold, which also is found in the copper metallochaperone Atx1.97 In addition to the metal-binding domain of the protein, UreE contains a second domain (shown in purple brackets) that resembles a peptide-binding domain of yeast Hsp40 or Sis1.98 To test whether this domain might function in peptide binding as part of the nickel delivery process, mutants in this region of the protein were examined. Notably, the loss of this entire domain along with the C-terminus fails to eliminate its ability to function in urease activation.63
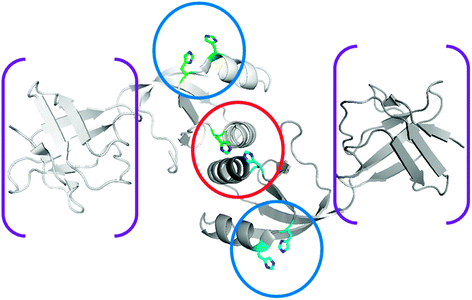 | ||
| Fig. 6 Structure of UreE. The structure of homodimeric K. aerogenes H144* UreE (PDB access code 1gmw), a truncated protein missing the C-terminal His-rich sequence, is depicted without metal ions. The central metal binding site (encircled in red) occurs at the subunit interface site and binds two nickel ions using, in part, His 96 residues from each subunit. Auxiliary metal binding sites (encircled in blue), involving the non-essential His 110 and His 112 residues, are presumed to donate nickel ions to the central site. When present, the C-terminal extension is also well positioned to provide nickel ions to the interfacial site. In addition to the metal-binding domain, a “peptide-binding domain” is present (in brackets) but is not essential for UreE function. | ||
UreE proteins have been purified and characterized to various extents from several other bacteria. P. mirabilis UreE contains a shorter His-rich C-terminus (HHHHDHHH) and also is dimeric.99 The H. pylori UreE protein, which lacks a His-rich region at its C-terminus, binds one nickel ion per dimer.100 More extensive investigations have been carried out on UreE of B. pasteurii, which binds two nickel ions per dimer.101–103 Significantly, the crystal structure of the zinc-bound protein is known,104 but like K. aerogenes UreE the nickel-bound structure is not available. The overall fold of B. pasteurii UreE is essentially identical to that from K. aerogenes, shown in Fig. 6. The B. pasteuriiprotein lacks residues corresponding to His 110 and His 112, and thus the periphery sites are absent; however, the residue corresponding to His 96 of K. aerogenes is conserved and this protein binds a single zinc atom at this interface site. Calorimetry studies provide confirmatory evidence that two nickel ions bind to this protein in an entropically-driven process (differing from the situation with K. aerogenes UreE),95 and results from XAS studies are compatible with Ni–Ni scattering from metal ions separated by 3.4 Å.102
The interaction of UreE with the UreABC–UreDFG complex is of great interest as this most likely represents the physiologically relevant activation complex that produces fully active urease. Significantly, incubation of UreABC–UreDFG plus purified UreE with physiologically relevant concentrations of bicarbonate, nickel, and GTP activates the ureaseapoprotein to wild-type levels.105E. coli cells expressing an intact K. aerogenesureasegene cluster encoding a UreB variant (involving the proposed hinge region mentioned above) exhibit deficiencies in urease activation and appear to have trapped some of the ureaseapoprotein in a UreABC–UreDFG–UreE complex.71 UreE also directly interacts with UreG according to analyses of the H. pylori system using yeast two-hybrid approaches.67,72
4.7 Other accessory proteins
Superimposed on the already complex urease activation scheme shown in Fig. 5, some bacteria possess additional accessory proteins encoded within their ureasegene clusters. As mentioned in section 4.1, genes encoding a proton-gated urea channel58 and suspected nickel ion permeases or multi-component, nickel-specific, ATP-binding cassette-type transporters have been reported.54,55 Consistent with a function in nickel transport, E. coli cells containing the putative A. pleuropneumoniae transporter genes (cbiKLMQ) along with this organism’s ureasegenes are urease positive when grown in Luria-Bertani medium, whereas cells containing only the ureasegenes require supplementation by nickel ions to achieve full urease activity.55Yersinia pseudotuberculosis also possesses a five-gene cluster (yntABCDE) located 5’ of the ureasegenes, and deletion of this region within the endogenous host abolishes urease activity and reduces the rates of nickel uptake by the microorganism.106Y. pseudotuberculosis also contains a gene, ureH located 3’ of the ureasegenes, that is related in sequence to nickel-cobalt transporter proteins; deletion of ureH leads to diminished rates of nickel uptake, confirming a function in nickel transport.106 In a similar manner, Streptococcus salivarius 57.I contains three genes (ureMQO) immediately downstream of the ureasegene cluster, and insertional inactivation of ureM leads to the inability to accumulate nickel ions and the consequent loss of urease activity.107Urease activation in H. pylori and related bacteria is further complicated by the involvement of several additional genes that are not adjacent to the ureasegene cluster. For example, the nixAgene encodes a nickel ion transporter according to uptake studies in recombinant E. coli cells containing this gene, and its presence along with the H. pyloriureasegenes significantly enhances urease activity in this heterologous host.108 Furthermore, mutagenesis of nixA in H. pylori reduces nickel ion transport and decreases urease activity.109 In addition to NixA, H. pylori contains the AbcABCD nickel ABC-type transporter.171 This microorganism also possesses an outer membrane transporter, energized by the TonB/ExbB/ExbD complex, that takes up nickel and enhances urease activity.110,111 Also of note are three cytoplasmic nickel-binding proteins: HspA, Hpn, and the Hpn-like protein. HspA is a GroES homologue containing a 27-residue C-terminal extension that is rich in His and Cys residues. In E. coli cells containing the H. pyloriureasegene cluster, co-expression of hspA stimulates the level of urease activity about four-fold.112,113 The extra domain binds two nickel ions and provides some protection against toxic levels of this metal, and it also binds two bismuth ions, a component of several anti-ulcer drugs.113,114 The Hpn protein is a 60-residue peptide containing 28 His residues.115 Deletion of the gene encoding Hpn within H. pylori either has no effect on urease activity115 or actually increases urease activity.116 This metal-binding protein117 is hypothesized to play a role in nickel storage that could be utilized for nickel donation while also functioning in metal detoxification.116,118 The Hpn-like protein, a 75-residue peptide containing 19 His and 30 Gln, binds two nickel ions per monomer.119 Deletion of the gene leads to an increase in urease activity while also increasing the sensitivity of cells to nickel, cobalt, and cadmium toxicity.116
Of greater interest to urease activation are two genes, hypA and hypB typically associated with hydrogenase biosynthesis in bacteria, that are required for full urease activity in H. pylori and H. hepaticus.120,121H. pylori contains a full complement of structural and accessory genes for the production of a [NiFe] hydrogenase, an enzyme that serves as an energy source for this bacterium.122 HypA is a nickel-binding protein that is proposed to be the nickel chaperone for hydrogenase maturation.123 In contrast, H. pylori HypB does not bind nickel, but this dimeric protein, which is homologous to UreG, possesses weak GTPase activity needed for hydrogenase activation.123 Furthermore, chemical cross-linking studies demonstrate that HypA and HypB can form a heterodimer. In both Helicobacter species mentioned above, mutations in hypA and hypB nearly abolish urease and hydrogenase activity.120,121 The mechanism responsible for the dual functioning of these activation proteins remains unclear, and neither HypA nor HypB are needed for heterologous activation of H. pyloriurease in E. coli.124 A possible hint of the site of interaction is the HypA–UreE heterodimer complex captured by chemical cross-linking studies, consistent with an interaction between these two proteins in the cell.125 Furthermore, tandem affinity purification experiments demonstrate interactions between HypB and UreG as well as HypB and urease.88 Further efforts are needed to define the interplay of these two activation systems in this microorganism.
5.0 In vivo maturation of eukaryotic ureases
As detailed in the preceding section 4.0, the mechanism of urease activation has been extensively investigated in the case of bacterial ureases; however, many fungi and plants also possess this enzyme which must undergo analogous activation processes. Genetic and biochemical studies related to urease activation in eukaryotes are summarized below.Fungal ureases contain a single type of subunit, but genetic studies reveal that multiple genes are needed for urease expression. For example, early studies with Neurospora crassa,126–128Aspergillus nidulans,129,130 and Schizosaccharomyces pombe,131 show that four distinct loci are required for obtaining active enzyme . The best studied fungal urease system is that in the fission yeast S. pombe where the enzyme was purified,132 the structural gene (ure2) identified,133 and candidate genes encoding UreD (ure4), UreF (ure3), and UreG (ure1) were identified.134 Of interest, the fungal UreG has 61% sequence identity to soybean UreG (see below) including the presence of a His-rich N-terminus that is not found in bacterial UreG sequences. The fungal UreF protein of S. pombe shares only 20% sequence identity with the soybean protein, but the plant gene rescues the corresponding yeast mutant, whereas this situation is not observed for UreD where the proteins share 30% sequence identity.134
Plants can possess multiple ureaseisozymes as well as genes encoding several maturation proteins. The historically interesting jack bean (C. ensiformis) system includes two structural ureasegenes,135,136 but no accessory genes have yet been reported. The better-studied soybean (Glycine max) system also has two ureaseisozymes : an embryo-specific form encoded at the Eu1 locus137,138 and a ubiquitously-expressed species encoded at the Eu4 locus.139,140 In addition, this plant contains several demonstrated urease accessory genes. For example, soybean UreD and UreF are orthologues of the bacterial and fungal genes; in the latter case, the plant gene complements a mutant involving the corresponding gene in S. pombe.134 The Eu3 locus was long known to exhibit pleiotropic effects on both ureases,141 and more recently was shown to encode a protein related to bacterial UreG proteins with a conserved P-loop motif and an added His-rich N-terminus that resembles the C-terminus of some bacterial UreE proteins and is probably involved in nickel binding.142Eu2 encodes another protein necessary for activation of both ureases,141 but its sequence and function remain unknown. Homologues encoding UreD-, UreF-, and UreG-like proteins are now known to exist in many other plant species, including tomato (Lycopersicon esculentum), potato (Solanum tuberosum), and Arabidopsis thaliana.134,143,144 Of interest, potato UreG complements a K. aerogenes ureG mutation143 and insertions into each of the three accessory genes of A. thaliana abolished urease activity.144
6.0 Regulation of urease by metal ions or other effectors
Ureolytic organisms utilize a wide variety of regulatory mechanisms to control urease. In bacterial pathogens that infect the urinary tract, such as P. mirabilis or P. stuartii, the AraC-like transcriptional activator UreR binds urea as an effector molecule and induces urease expression.145,146 A pH-dependent regulatory mechanism operates through cis-acting elements in the dental plaque microorganism S. salivarius.147,148 In some environmental isolates that utilize urea as a nitrogen source, the NtrC transcriptional activator binds to the urease promoter and allows for nitrogen-dependent regulation of urease.149 A cascade version of this mechanism is utilized by K. aerogenes which uses NtrC to control levels of the nitrogen assimilation control protein that then directly regulates urease expression.150,151 Three factors, CodY, GlnR, and Spo0H, all regulate urease expression in response to nitrogen availability in the spore-forming bacterium B. subtilis.152 In contrast, urease of B. pasteurii appears to be nearly constitutively expressed at high levels.153 Additional levels of complexity are known in eukaryotes. As an illustration, the algal partner in the lichen Evernia prunastri is suggested to produce a protein that inhibits urease synthesis in the associated fungus.154 As another example, soybean regulates its isozymes in a temporal and tissue-specific manner.139 Cucumber leaf urease is reported to be induced by cobalt ions, although the molecular basis of this regulation is unknown.155 These and other regulatory mechanisms will not be further described here; rather, this section focuses on metal-dependent regulation of bacterial ureases.The clearest example of urease expression being regulated by metal ions has been described in H. pylori. In particular, both Northern hybridization studies and analysis of β-galactosidase activity in a ureA::lacZ transcriptional fusion show that supplementation of brucella growth medium with nickel ions leads to an increase (up to 3.5-fold) in transcription of the ureasegenes.156 Notably, the effects are not observed upon addition of cadmium, cobalt, copper, iron, manganese, or zinc, but are specific to nickel. This same report demonstrates a two-fold effect of a fur mutant (lacking a functional ferric uptake regulator) in the nickel-supplemented cells, suggesting that Fur also modulates the levels of urease. Additional evidence supporting nickel-dependent regulation of urease activity in H. pylori is derived from studies of a nikR mutant, defective in the corresponding nickel regulatory protein.157 The mutant cells lack the ability to increase urease expression in the presence of nickel. The same phenotype is noted when a 19-bp palindromic sequence just upstream of the ureaseoperon is removed. Furthermore, the nikR mutant cells are growth inhibited by nickel ions at concentrations greater than 100 μM whereas the wild-type cells tolerate high concentrations of nickel ions. These results are consistent with nickel-bound NikR acting as an activator of urease and a repressor of nickel transport. These findings were extended to demonstrate that metal-bound NikR activates or represses several genes (ureA, nixA, copA2, hpn, and hpn-like, several of which are known to encode proteins that bind nickel ions) while repressing a large number of other genes that include nickel uptake factors and fur.158,159,162,172 Crystallographic studies have elucidated the structure of H. pylori NikR in the apoprotein and nickel-bound states.160 An elegant model was described for the pH-dependent modulation of the NikR-dependent regulation, accounting for acid adaptation of H. pylori.161 Additional studies by several laboratories have characterized the binding of nickel to NikR and its interaction with the urease promoter in this microorganism.162–169
Studies carried out with three other species of Helicobacter (i.e., H. mustelae, H. acinonychis, and H. felis) highlight another intriguing aspect of metal-dependent urease regulation. These microorganisms possess two sets of ureasegenes, one which contains the complete ureasegene cluster and one containing only a second copy of the structural genes (termed ureA2 and ureB2).60,170 In each case, the second copy of each subunit is closely related (more than 50% identical) to the first copy. In the best studied case of H. mustelae, quantitative RT-PCR was used to show that ureAB is induced by nickel ions whereas ureA2B2 is inversely regulated by nickel and increases with iron supplementation.60 Immunological results confirm this inverse regulation of the two proteins in H. mustelae and demonstrate the same situation exists in the other two species. A nikR/ureB mutant exhibits small amounts of urease activity, consistent with ureA2B2 encoding a functional enzyme that does not utilize nickel ions. Furthermore, a nikR/ureB/ureG mutant possesses the same activity levels, suggesting that accessory proteins are not required for synthesis of an active form of the second urease. Similarly, hypB was shown not to be required for this activity. In contrast to the stability observed for most ureases, the activity associated with UreA2B2 is lost upon disruption of the cells. These results led the researchers to put forth the intriguing hypothesis that the second urease may be an oxygen-sensitive, iron-containing urease. Congruent with this hypothesis, these Helicobacter species are noted as being associated with carnivores whose food source is depleted in nickel, but rich in iron. Studies of the second enzyme from H. mustelae are under way in an effort to characterize this potential iron-dependent urease.
7.0 Perspectives
Despite extensive efforts to understand the catalysis, nickel dependence, and molecular activation mechanism of urease, many fundamental questions remain unanswered.Structural, biochemical, and computational data provide us with a wealth of detail about the urease active site; nevertheless, as illustrated in Fig. 3, the precise reactions leading to the final release of carbamic acid and ammonia are unclear. The chemistries inherent to these reactions are not restricted to nickel, yet all ureases characterized to date possess nickel. Do non-nickel forms of this enzyme exist in nature? The generation of partially active urease by treatment of apoprotein with bicarbonate plus manganese or cobalt (for C319A protein) lends support to this idea. In addition, the discovery that ureA2B2 of H. mustelae is inversely regulated by nickel and stimulated by iron further supports this notion.
The definitive roles of the UreD, UreE, UreF, and UreG accessory proteins during urease maturation remain elusive. We discussed earlier the notion that UreDFG, perhaps pre-formed, might serve as a GTP-dependent molecular chaperone while UreE functions as a metallochaperone, but this proposal demands further verification. Using MBP-UreD or other soluble forms of UreD, we anticipate that biochemical and structural analyses will be carried out on this yet uncharacterized protein. Such studies, combined with follow-up investigations of UreABC–UreD, may shed light on the ability of ureD to increase the in vitro activation competence of the bound apoprotein. Additional biochemical and structural studies will be performed with the UreEF fusion protein and other soluble forms of UreF, as well as with the UreABC–UreDF complex. This effort should provide new insight into the structure of UreF and its function in the maturation process. The postulated similarity of UreF to known GTPase activating factors is intriguing as this protein may act in concert with UreG (a known GTPase) in the UreABC–UreDFG activation complex to couple the energy of GTP hydrolysis for efficient urease activation. The structure of UreG still needs to be determined, its metal-binding properties more clearly examined, and its interaction with other urease components investigated at greater resolution. Our current model designates UreE as responsible for nickel delivery; however, the mechanism of delivery is unknown. For example, is nickel transferred directly from UreE to the structural subunits or do other components function as intermediaries in this process? How GTP hydrolysis drives this process is also uncertain.
The findings that additional non-contiguous genes are often encoded near ureasegene clusters and the recent discovery that hydrogenase maturation factors affect urease activation in at least two strains of Helicobacter are also of interest. The presence of urea and nickel transport/assimilation systems linked to urease activity in other organisms and the lack of accessory proteins in B. subtilis demonstrate that urease maturation may be influenced by other cellular factors. The convergence of two maturation pathways may increase the efficiency of metallocenter assembly by coordinating the delivery of nickel (using HypA and UreE) or facilitating the nucleotide dependence (using HypB and UreG).
Relatively little is known about urease maturation in eukaryotic systems. Parallels between the fungal and plant activation components with the bacterial systems need to be further explored, and novel features, such as the product of Eu2 in soybean, require additional investigation. Studies to uncover cellular urea and nickel homeostasis systems and to explore novel urease maturation pathways in both eukaryotes and prokaryotes will surely lead to new discoveries in urease biology.
Acknowledgements
Studies related to this topic in the Hausinger laboratory are supported by NIH grant DK04586.References
- J. B. Sumner, J. Biol. Chem., 1926, 69, 435–441 CAS.
- N. E. Dixon, C. Gazzola, R. L. Blakeley and B. Zerner, J. Am. Chem. Soc., 1975, 97, 4131–4133 CrossRef CAS.
- H. L. T. Mobley, M. D. Island and R. P. Hausinger, Microbiol. Rev., 1995, 59, 451–480 CAS.
- H. L. T. Mobley and R. P. Hausinger, Microbiol. Rev., 1989, 53, 85–108 CAS.
- R. J. C. McLean, J. C. Nickel, K.-J. Cheng and J. W. Costerton, CRC Crit. Rev. Microbiol., 1988, 16, 37–79 CrossRef CAS.
- C. M. Collins and S. E. F. D'Orazio, Mol. Microbiol., 1993, 9, 907–913 CrossRef CAS.
- J. C. Atherton, Annu. Rev. Pathol. Mech. Dis., 2006, 1, 63–96 Search PubMed.
- J. G. Kusters, A. H. M. Van Vliet and E. J. Kuipers, Clin. Microbiol. Rev., 2006, 19, 449–490 CrossRef CAS.
- K. Stingl, K. Altendorf and E. P. Bakker, Trends Microbiol., 2002, 10, 70–74 CrossRef CAS.
- L. E. Zonia, N. E. Stebbins and J. C. Polacco, Plant Physiol., 1995, 107, 1097–1103 CrossRef CAS.
- R. P. Hausinger, in Biochemistry of Nickel, Plenum Publishing Corp., New York, 1993, pp. 23–57 Search PubMed.
- R. P. Hausinger and P. A. Karplus, in Handbook of Metalloproteins, ed. K. Wieghardt, R. Huber, T. L. Poulos and A. Messerschmidt, John Wiley & Sons Ltd., West Sussex, UK, 2001, pp. 867–879 Search PubMed.
- C. Follmer, Phytochemistry, 2008, 69, 18–28 CrossRef CAS.
- S. Ciurli, in Metal Ions in Life Sciences, ed. A. Sigel, H. Sigel and R. K. O. Sigel, John Wiley & Sons, Ltd., Chichester, UK, 2007, vol. 2, pp. 241–278 Search PubMed.
- E. Jabri, M. B. Carr, R. P. Hausinger and P. A. Karplus, Science, 1995, 268, 998–1004 CrossRef CAS.
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Miletti, S. Ciurli and S. Mangani, Structure, 1999, 7, 205–216 CrossRef CAS.
- N.-C. Ha, S.-T. Oh, J. Y. Sung, K.-A. Cha, M. H. Lee and B.-H. Oh, Nat. Struct. Biol., 2001, 8, 505–509 CrossRef CAS.
- L. Sheridan, C. M. Wilmont, K. D. Cromie, P. van der Logt and S. E. V. Phillips, Acta Crystallogr., Sect. D, 2002, D58, 374–376 CrossRef CAS.
- M. A. Pearson, L. O. Michel, R. P. Hausinger and P. A. Karplus, Biochemistry, 1997, 36, 8164–8172 CrossRef CAS.
- L. Alagna, S. S. Hasnain, B. Piggott and D. J. Williams, Biochem. J., 1984, 220, 591–595 CAS.
- P. A. Clark, D. E. Wilcox and R. A. Scott, Inorg. Chem., 1990, 29, 579–581 CrossRef CAS.
- S. Wang, M. H. Lee, R. P. Hausinger, P. A. Clark, D. E. Wilcox and R. A. Scott, Inorg. Chem., 1994, 33, 1589–1593 CrossRef CAS.
- S. Benini, S. Ciurli, H. F. Nolting and S. Mangani, Eur. J. Biochem., 1996, 239, 61–66 CrossRef CAS.
- R. L. Blakeley, N. E. Dixon and B. Zerner, Biochim. Biophys. Acta, 1983, 744, 219–229 CAS.
- M. J. Todd and R. P. Hausinger, J. Biol. Chem., 1989, 264, 15835–15842 CAS.
- M. G. Finnegan, A. Kowal, M. T. Werth, P. A. Clark, D. E. Wilcox and M. K. Johnson, J. Am. Chem. Soc., 1991, 113, 4030–4032 CrossRef CAS.
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Ciurli and S. Mangani, J. Biol. Inorg. Chem., 1998, 3, 268–273 CrossRef CAS.
- P. A. Clark and D. E. Wilcox, Inorg. Chem., 1989, 28, 1326–1333 CrossRef CAS.
- I.-S. Park, L. O. Michel, M. A. Pearson, E. Jabri, P. A. Karplus, S. Wang, J. Dong, R. A. Scott, B. P. Koehler, M. K. Johnson and R. P. Hausinger, J. Biol. Chem., 1996, 271, 18632–18637 CrossRef CAS.
- M. A. Pearson, R. A. Schaller, L. O. Michel, P. A. Karplus and R. P. Hausinger, Biochemistry, 1998, 37, 6214–6220 CrossRef CAS.
- M. A. Pearson, I.-S. Park, R. A. Schaller, L. O. Michel, P. A. Karplus and R. P. Hausinger, Biochemistry, 2000, 39, 8575–8584 CrossRef CAS.
- N. E. Dixon, P. W. Riddles, C. Gazzola, R. L. Blakeley and B. Zerner, Can. J. Biochem., 1980, 58, 1335–1344 CAS.
- P. A. Karplus, M. A. Pearson and R. P. Hausinger, Acc. Chem. Res., 1997, 30, 330–337 CrossRef CAS.
- R. K. Andrews, A. Dexter, R. L. Blakeley and B. Zerner, J. Am. Chem. Soc., 1986, 108, 7124–7125 CrossRef CAS.
- M. J. Todd and R. P. Hausinger, Biochemistry, 2000, 39, 5389–5396 CrossRef CAS.
- K. Yamaguchi, S. Koshino, F. Akagi, M. Suzuki, A. Uehara and S. Suzuki, J. Am. Chem. Soc., 1997, 119, 5752–5753 CrossRef CAS.
- F. Meyer, E. Kaifer, P. Kircher, K. Heinze and H. Pritzkow, Chem.–Eur. J., 1999, 5, 1617–1630 CrossRef CAS.
- A. M. Barrios and S. J. Lippard, J. Am. Chem. Soc., 2000, 122, 9172–9177 CrossRef CAS.
- F. Musiani, E. Arnofi, R. Casadio and S. Ciurli, J. Biol. Inorg. Chem., 2001, 6, 300–314 CrossRef CAS.
- D. Suárez, N. Diaz and K. M. Merz, Jr, J. Am. Chem. Soc., 2003, 125, 15324–15337 CrossRef CAS.
- G. Estiu and K. M. Merz, Jr, J. Am. Chem. Soc., 2004, 126, 11832–11842 CrossRef CAS.
- G. Estiu and K. M. Merz, Jr, Biochemistry, 2006, 45, 4429–4443 CrossRef CAS.
- G. Estiu and K. M. Merz, Jr, J. Phys. Chem. B, 2007, 111, 10263–10274 CrossRef CAS.
- I.-S. Park and R. P. Hausinger, Science, 1995, 267, 1156–1158 CAS.
- I.-S. Park and R. P. Hausinger, Biochemistry, 1996, 35, 5345–5352 CrossRef CAS.
- K. Yamaguchi, N. J. Cosper, C. Stalhanske, R. A. Scott, M. A. Pearson, P. A. Karplus and R. P. Hausinger, J. Biol. Inorg. Chem., 1999, 4, 468–477 CrossRef CAS.
- K. Yamaguchi and R. P. Hausinger, Biochemistry, 1997, 36, 15118–15122 CrossRef CAS.
- G. J. King and B. Zerner, Inorg. Chim. Acta, 1997, 255, 381–388 CrossRef CAS.
- R. P. Hausinger, G. J. Colpas and A. Soriano, ASM News, 2001, 67, 78–84 Search PubMed.
- S. B. Mulrooney and R. P. Hausinger, FEMS Microbiol. Rev., 2003, 27, 239–261 CrossRef CAS.
- M. H. Lee, S. B. Mulrooney, M. J. Renner, Y. Markowicz and R. P. Hausinger, J. Bacteriol., 1992, 174, 4324–4330 CAS.
- S. B. Mulrooney and R. P. Hausinger, J. Bacteriol., 1990, 172, 5837–5843 CAS.
- A. Toffanin, E. Cadahia, J. Imperial, T. Ruiz-Argüeso and J. M. Palacios, Arch. Microbiol., 2002, 177, 290–298 CrossRef CAS.
- M. Maeda, M. Hidaka, A. Nakamura, H. Masaki and T. Uozumi, J. Bacteriol., 1994, 176, 432–442 CAS.
- J. T. Bossè, H. D. Gilmour and J. I. MacInnes, J. Bacteriol., 2001, 183, 1242–1247 CrossRef CAS.
- C. L. Clayton, M. J. Pallen, H. Kleanthous, B. W. Wren and S. Tabaqchali, Nucleic Acids Res., 1990, 18, 362 CrossRef CAS.
- A. Labigne, V. Cussac and P. Courcoux, J. Bacteriol., 1991, 173, 1920–1931 CAS.
- D. L. Weeks, S. Eskandar, D. R. Scott and G. Sachs, Science, 2000, 287, 482–485 CrossRef CAS.
- V. Cussac, R. L. Ferrero and A. Labigne, J. Bacteriol., 1992, 174, 2466–2473 CAS.
- J. Stoof, S. Breijer, R. G. J. Pot, D. van der Neut, E. J. Kuipers and A. H. M. van Vliet, Environ. Microbiol., 2008, 10, 2586–2597 CrossRef CAS.
- J. K. Kim, S. B. Mulrooney and R. P. Hausinger, J. Bacteriol., 2005, 187, 7150–7154 CrossRef CAS.
- S. B. Mulrooney, H. S. Pankratz and R. P. Hausinger, J. Gen. Microbiol., 1989, 135, 1769–1776 CAS.
- S. B. Mulrooney, S. K. Ward and R. P. Hausinger, J. Bacteriol., 2005, 187, 3581–3585 CrossRef CAS.
- S. E. Walz, S. K. Wray, S. I. Hull and R. E. Hull, J. Bacteriol., 1988, 170, 1027–1033 CAS.
- G.-F. Gerlach, S. Clegg and W. A. Nichols, FEMS Microbiol. Lett., 1988, 50, 131–135 CrossRef CAS.
- S. B. Mulrooney, M. J. Lynch, H. L. T. Mobley and R. P. Hausinger, J. Bacteriol., 1988, 170, 2202–2207 CAS.
- P. Voland, D. L. Weeks, E. A. Marcus, C. Prinz, G. Sachs and D. Scott, Liver Physiol., 2003, 284, G96–G106 Search PubMed.
- J.-U. Park, J.-Y. Song, Y.-C. Kwon, M.-J. Chung, J.-S. Jun, J.-W. Park, S.-G. Park, H.-R. Hwang, S.-H. Choi, S.-C. Baik, H.-L. Kang, H.-S. Youn, W.-K. Lee, M.-J. Cho and K.-H. Rhee, Mol. Cell., 2005, 20, 371–377 CAS.
- I.-S. Park, M. B. Carr and R. P. Hausinger, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3233–3237 CrossRef CAS.
- Z. Chang, J. Kuchar and R. P. Hausinger, J. Biol. Chem., 2004, 279, 15305–15313 CrossRef CAS.
- S. Quiroz-Valenzuela, S. C. K. Sukuru, R. P. Hausinger, L. A. Kuhn and W. T. Heller, Arch. Biochem. Biophys., 2008, 480, 51–57 CrossRef CAS.
- J.-C. Rain, L. Selig, H. de Reuse, V. Battaglia, C. Reverdy, S. Simon, G. Lenzen, F. Petel, J. Wojcik, V. Schächter, Y. Chemama, A. Labigne and P. Legrain, Nature, 2001, 409, 211–215 CrossRef CAS.
- S. R. Heimer and H. L. Mobley, J. Bacteriol., 2001, 183, 1423–1433 CrossRef CAS.
- M. B. C. Moncrief and R. P. Hausinger, J. Bacteriol., 1996, 178, 5417–5421 CAS.
- K. Y. Kim, C. H. Yang and M. H. Lee, Arch. Pharm. Res., 1999, 22, 274–278 Search PubMed.
- J. K. Kim and R. P. Hausinger, J. Bacteriol., 2006, 188, 8413–8420 CrossRef CAS.
- M. Salomone-Stagni, B. Zambelli, F. Musiani and S. Ciurli, Proteins, 2007, 68, 749–761 Search PubMed.
- M. B. C. Moncrief and R. P. Hausinger, J. Bacteriol., 1997, 179, 4081–4086 CAS.
- B. Zambelli, M. Stola, F. Musiani, K. De Vriendt, B. Samyn, B. Devreese, J. Van Beeumen, A. Dikiy, D. A. Bryant and S. Ciurli, J. Biol. Chem., 2005, 280, 4684–4695 CAS.
- B. Zambelli, F. Musiani, M. Savini, P. Tucker and S. Ciurli, Biochemistry, 2007, 46, 3171–3182 CrossRef CAS.
- B. Zambelli, P. Turano, F. Musiani, P. Neyroz and S. Ciurli, Proteins, 2008, 74, 222–239 Search PubMed.
- N. Mehta, S. Benoit and R. J. Maier, Microb. Pathog., 2003, 35, 229–234 CrossRef CAS.
- P. Neyroz, B. Zambelli and S. Ciurli, Biochemistry, 2006, 45, 8918–8930 CrossRef CAS.
- R. Gasper, A. Scrima and A. Wittinghofer, J. Biol. Chem., 2006, 281, 27492–27502 CrossRef CAS.
- I.-S. Park and R. P. Hausinger, J. Bacteriol., 1995, 177, 1947–1951 CAS.
- A. Soriano and R. P. Hausinger, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11140–11144 CrossRef CAS.
- S. Quiroz, J. K. Kim, S. B. Mulrooney and R. P. Hausinger, in Metal Ions in Life Sciences, ed. A. Sigel, H. Sigel and R. K. O. Sigel, John Wiley & Sons, New York, 2007, vol. 2, pp. 519–544 Search PubMed.
- K. Stingl, K. Schauer, C. Ecobichon, A. Labigne, P. Lenormand, J.-C. Rousselle, A. Namane and H. de Reuse, Mol. Cell. Proteom., 2008, 7, 2429–2441 CrossRef CAS.
- G. J. Colpas, T. G. Brayman, L.-J. Ming and R. P. Hausinger, Biochemistry, 1999, 38, 4078–4088 CrossRef CAS.
- F. Musiani, B. Zambelli, M. Stola and S. Ciurli, J. Inorg. Biochem., 2004, 98, 803–813 CrossRef CAS.
- M. H. Lee, H. S. Pankratz, S. Wang, R. A. Scott, M. G. Finnegan, M. K. Johnson, J. A. Ippolito, D. W. Christianson and R. P. Hausinger, Protein Sci., 1993, 2, 1042–1052 CrossRef CAS.
- T. G. Brayman and R. P. Hausinger, J. Bacteriol., 1996, 178, 5410–5416 CAS.
- G. J. Colpas, T. G. Brayman, J. McCracken, M. A. Pressler, G. T. Babcock, L.-J. Ming, C. M. Colangelo, R. A. Scott and R. P. Hausinger, J. Biol. Inorg. Chem., 1998, 3, 150–160 CrossRef CAS.
- G. J. Colpas and R. P. Hausinger, J. Biol. Chem., 2000, 275, 10731–10737 CrossRef CAS.
- N. E. Grossoehme, S. B. Mulrooney, R. P. Hausinger and D. E. Wilcox, Biochemistry, 2007, 46, 10506–10516 CrossRef CAS.
- H. K. Song, S. B. Mulrooney, R. Huber and R. P. Hausinger, J. Biol. Chem., 2001, 276, 49359–49364 CrossRef CAS.
- A. C. Rosenzweig, D. L. Huffman, M. Y. Hou, A. K. Wernimont, R. A. Pufahl and T. V. O'Halloran, Structure, 1999, 7, 605–617 CrossRef.
- B. Sha, S.-K. Lee and D. M. Cyr, Structure, 2000, 8, 799–807 CrossRef CAS.
- B. Sriwanthana, M. D. Island, D. Maneval and H. L. T. Mobley, J. Bacteriol., 1994, 176, 6836–6841 CAS.
- S. Benoit and R. J. Maier, J. Bacteriol., 2003, 185, 4787–4795 CrossRef CAS.
- S. Ciurli, N. Safarof, S. Miletti, A. Dikiy, S. K. Christensen, K. Kornetzky, D. A. Bryant, I. Vandenberghe, B. Devreese, B. Samyn, H. Remaut and J. Van Beeumen, J. Biol. Inorg. Chem., 2002, 7, 623–631 CAS.
- M. Stola, F. Musiani, S. Mangani, P. Turano, N. Safarov, B. Zambelli and S. Ciurli, Biochemistry, 2006, 45, 6495–6509 CrossRef CAS.
- H.-S. Won, Y.-H. Lee, J.-H. Kim, I. S. Shin, M. H. Lee and B.-J. Lee, J. Biol. Chem., 2004, 279, 17466–17472 CrossRef CAS.
- H. Remaut, N. Safarof, S. Ciurli and J. Van Beeumen, J. Biol. Chem., 2001, 276, 49365–49370 CrossRef CAS.
- A. Soriano, G. J. Colpas and R. P. Hausinger, Biochemistry, 2000, 39, 12435–12440 CrossRef CAS.
- F. Sebbane, M. A. Mandrand-Bethelot and M. Simonet, J. Bacteriol., 2002, 184, 5706–5713 CrossRef CAS.
- Y.-Y. M. Chen and R. A. Burne, J. Bacteriol., 2003, 185, 6773–6779 CrossRef CAS.
- H. L. T. Mobley, R. M. Garner and P. Bauerfeind, Mol. Microbiol., 1995, 16, 97–109 CrossRef CAS.
- P. Bauerfiend, R. M. Garner and H. L. T. Mobley, Infect. Immun., 1996, 64, 2877–2880.
- G. S. Davis, E. L. Flannery and H. L. T. Mobley, Infect. Immun., 2006, 74, 6811–6820 CrossRef CAS.
- K. Schauer, B. Gouget, M. Carrière, A. Labigne and H. de Reuse, Mol. Microbiol., 2007, 63, 1054–1068 CrossRef CAS.
- S. Suerbaum, J.-M. Thiberge, I. Kansau, R. L. Ferrero and A. Labigne, Mol. Microbiol., 1994, 14, 959–974 CrossRef CAS.
- I. Kansau, F. Guillain, J.-M. Thiberge and A. Labigne, Mol. Microbiol., 1996, 22, 1013–1023 CrossRef CAS.
- S. Cun, H. Li, R. Ge, M. C. M. Lin and H. Sun, J. Biol. Chem., 2008, 283, 15142–15151 CrossRef CAS.
- J. V. Gilbert, J. Ramakrishna, F. W. Sunderman Jr, A. Wright and A. G. Plaut, Infect. Immun., 1995, 63, 2682–2688 CAS.
- S. Seshadri, S. L. Benoit and R. J. Maier, J. Bacteriol., 2007, 189, 4120–4126 CrossRef CAS.
- R. Ge, Y. Zhang, X. Sun, R. M. Watt, Q.-Y. He, J.-D. Huang, D. E. Wilcox and H. Sun, J. Am. Chem. Soc., 2006, 128, 11330–11331 CrossRef CAS.
- R. Ge, R. M. Watt, X. Sun, J. A. Tanner, Q.-Y. He, J.-D. Huang and H. Sun, Biochem. J., 2006, 393, 285–293 CrossRef CAS.
- Y.-B. Z. Zeng, D.-M. H. Li and H. Sun, J. Biol. Inorg. Chem., 2008, 13, 1121–1131 CrossRef CAS.
- J. W. Olson, N. S. Mehta and R. J. Maier, Mol. Microbiol., 2001, 39, 176 CrossRef CAS.
- S. L. Benoit, A. L. Zbell and R. J. Maier, Microbiology, 2007, 153, 3748–3756 CrossRef CAS.
- J. W. Olson and R. J. Maier, Science, 2002, 298, 1788–1790 CrossRef CAS.
- N. S. Mehta, J. W. Olson and R. J. Maier, J. Bacteriol., 2003, 185, 726–734 CrossRef CAS.
- L.-T. Hu and H. L. T. Mobley, Infect. Immun., 1993, 61, 2563–2569 CAS.
- S. L. Benoit, N. Mehta, M. V. Weinberg, C. Maier and R. J. Maier, Microbiology, 2007, 153, 1474–1482 CrossRef CAS.
- H. G. Kolmark, Mutat. Res., 1969, 8, 51–63 CrossRef CAS.
- P. Haysman and H. B. Howe, Jr, Can. J. Genet. Cytol., 1971, 13, 256–269 Search PubMed.
- E. W. Benson and H. B. Howe, Jr, Mol. Gen. Genet., 1978, 165, 277–282 CrossRef CAS.
- E. M. Mackay and J. A. Pateman, J. Gen. Microbiol., 1980, 116, 249–251 CAS.
- E. M. Mackay and J. A. Pateman, Biochem. Genet., 1982, 20, 763–776 CrossRef CAS.
- J. R. Kinghorn and R. Fluri, Curr. Genet., 1984, 8, 99–105 CrossRef CAS.
- M. W. Lubbers, S. B. Rodriquez, N. K. Honey and R. J. Thornton, Can. J. Microbiol., 1996, 42, 132–140 CrossRef CAS.
- Y. Tange and O. Niwa, Curr. Genet., 1997, 32, 244–246 CrossRef CAS.
- M. Bacanamwo, C.-P. Witte, M. W. Lubbers and J. C. Polacco, Mol. Gen. Genet., 2002, 268, 525–534 CrossRef CAS.
- P. W. Riddles, V. Whan, R. L. Blakeley and B. Zerner, Gene, 1991, 108, 265–267 CrossRef CAS.
- M. Pires-Alves, Grossi-de-Sá, G. B. S. Barcellos, C. R. Carlini and M. G. Moraes, Plant Cell Physiol., 2003, 44, 139–145 CrossRef CAS.
- L. E. Meyer-Bothling and J. E. Polacco, Mol. Gen. Genet., 1987, 209, 439–444 CrossRef CAS.
- R. W. Krueger, M. A. Holland, D. Chisholm and J. C. Polacco, Gene, 1987, 54, 41–50 CrossRef CAS.
- M. A. Holland, J. D. Griffin, L. E. Meyer-Bothling and J. C. Polacco, Dev. Genet., 1987, 8, 375–387 CrossRef CAS.
- R. S. Torisky, J. D. Griffin, R. L. Yenofsky and J. E. Polacco, Mol. Gen. Genet., 1994, 242, 404–414 CAS.
- L. E. Meyer-Bothling, J. C. Polacco and S. R. Cianzio, Mol. Gen. Genet., 1987, 209, 432–438 CrossRef CAS.
- S. K. Freyermuth, M. Bacanamwo and J. C. Polacco, Plant J., 2000, 21, 53–60 CrossRef CAS.
- C.-P. Witte, E. Isadore, S. A. Tiller, H. V. Davies and M. A. Taylor, Plant Mol. Biol., 2001, 45, 169–179 CrossRef CAS.
- C.-P. Witte, M. G. Rosso and T. Romeis, Plant Physiol., 2005, 139, 1155–1162 CrossRef CAS.
- S. E. F. D’Orazio and C. M. Collins, J. Bacteriol., 1993, 175, 3459–3467 CAS.
- I. Gendlina, D. M. Gutman, V. Thomas and C. M. Collins, J. Biol. Chem., 2002, 277, 37349–37358 CrossRef CAS.
- C. H. Sisson, H. E. R. Perinpanayagam and E. M. Hancock, Oral Microbiol. Immunol., 1992, 7, 159–164.
- Y.-Y. M. Chen, M. J. Betzenhauser and R. A. Burne, Microbiology, 2002, 148, 3599–3608 CAS.
- B. Masepohl, B. Kaiser, N. Isakovic, C. L. Richard, R. G. Kranz and W. Klipp, J. Bacteriol., 2001, 183, 637–643 CrossRef CAS.
- A. Macaluso, E. A. Best and R. A. Bender, J. Bacteriol., 1990, 172, 7249–7255 CAS.
- Q. Liu and R. A. Bender, J. Bacteriol., 2007, 189, 7593–7599 CrossRef CAS.
- L. V. Wray, Jr, A. E. Ferson and S. H. Fisher, J. Bacteriol., 1997, 179, 5494–5501 CAS.
- A. D. Larson and R. E. Kallio, J. Bacteriol., 1954, 68, 67–73 CrossRef CAS.
- E. Perez-Urria, M. Rodriguez and C. Vicente, Plant Mol. Biol., 1989, 13, 665–672 CrossRef CAS.
- Y. Watanabe, T. Iizuka and N. Shimada, Soil Sci. Plant Nutr., 1994, 40, 545–548 Search PubMed.
- A. H. M. van Vleit, E. J. Kuipers, B. Waidner, B. J. Davies, N. de Vries, C. W. Penn, C. M. J. E. Vandenbroucke-Grauls, M. Kist, S. Bereswill and J. G. Kusters, Infect. Immun., 2001, 69, 4891–4897 CrossRef.
- A. H. M. van Vliet, S. W. Poppelaars, B. J. Davies, J. Stoof, S. Bereswill, M. Kist, C. W. Penn, E. J. Kuipers and J. G. Kusters, Infect. Immun., 2002, 70, 2846–2852 CrossRef.
- M. Contreras, J.-M. Thiberg, M.-A. Mandrand-Berthelot and A. Labigne, Mol. Microbiol., 2003, 49, 947–963 CrossRef CAS.
- F. D. Ernst, J. Stoof, W. M. Horrevoets, E. J. Kuipers, J. G. Kusters and A. H. M. Van Vliet, Infect. Immun., 2006, 74, 6821–6828 CrossRef CAS.
- C. Dian, K. Schauer, U. Kapp, S. M. McSweeney, A. Labigne and L. Terradot, J. Mol. Biol., 2006, 361, 715–730 CrossRef CAS.
- A. H. M. van Vliet, F. D. Ernst and J. G. Kusters, Trends Microbiol., 2004, 12, 489–494 CrossRef CAS.
- F. D. Ernst, E. J. Kuipers, A. Heijens, R. Sarwari, J. Stoof, C. W. Penn, J. G. Kusters and A. H. van Vliet, Infect Immun, 2005, 73, 7252–7258 CrossRef CAS.
- L. O. Abraham, Y. Li and D. B. Zamble, J. Inorg. Biochem., 2006, 100, 1005–1014 CrossRef CAS.
- C. Fauquant, R. E. Diederix, A. Rodrigue, C. Dian, U. Kapp, L. Terradot, M. A. Mandrand-Berthelot and I. Michaud-Soret, Biochimie, 2006, 88, 1693–1705 CrossRef CAS.
- N. S. Dosanjh, N. A. Hammerbacher and S. L. Michel, Biochemistry, 2007, 46, 2520–2529 CrossRef CAS.
- B. Zambelli, M. Belluci, A. Danielli, V. Scarlato and S. Ciurli, Chem. Commun., 2007, 3649–3651 RSC.
- E. L. Benati and P. T. Chivers, J. Biol. Chem., 2007, 282, 20365–20375 CrossRef.
- B. Zambelli, A. Danielli, S. Romagnoli, P. Neyroz, S. Ciurli and V. Scarlato, J. Mol. Biol., 2008, 383, 1129–1143 CrossRef CAS.
- N. S. Dosanjh, A. L. West and S. L. Michel, Biochemistry, 2009, 48, 527–536 CrossRef CAS.
- R. G. J. Pot, J. Stoof, P. J. M. Nuijten, L. A. M. de Haan, P. Loeffen, E. J. Kuipers, A. H. M. Van Vliet and J. G. Kusters, FEMS Immunol. Med. Microbiol., 2007, 50, 273–279 CrossRef CAS.
- J. K. Hendricks and H. L. Mobley, J. Bacteriol., 1997, 179, 5892–5902 CAS.
- L. Wolfram, E. Haas and P. Bauerfeind, J. Bacteriol., 2006, 188, 1245–1250 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |