Sol–gel synthesis and structural characterisation of P2O5–B2O3–Na2O glasses for biomedical applications
Daniela
Carta
*a,
Jonathan C.
Knowles
b,
Paul
Guerry
c,
Mark E.
Smith
c and
Robert J.
Newport
a
aSchool of Physical Sciences, University of Kent, Canterbury, CT2 7NR, UK. E-mail: dcarta@unica.it
bUCL Eastman Dental Institute, 256 Gray's Inn Road, London, WC1X 8LD, UK
cDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
First published on 10th November 2008
Abstract
Glasses in the system 40(P2O5)–x(B2O3)–(60 − x)(Na2O) (10 ≤ x ≤ 25 mol%) were prepared by the sol–gel technique. A mixture of mono- and diethylphosphates was used as precursor for P2O5, boric acid and sodium methoxide were used as source compounds for B2O3 and Na2O, respectively. The dried gels obtained were heat treated at 200, 300 and 400 °C. Structural development occurring during heat treatment and changes with composition were investigated using X-ray diffraction, thermal analysis, infrared spectroscopy, 11B and 31P solid state NMR. Systems with x = 20 and x = 25 mol% are amorphous up to 400 °C, whereas systems with lower B2O3 content are partially crystalline. This work extends sol–gel preparation of amorphous borophosphate systems having P2O5 as the main component.
1. Introduction
Borophosphates glasses have been an interesting subject of research in recent years for their applications in many different areas, e.g. as electrolytes in solid state electrochemical cells for fast ion conduction (alkali and silver borophosphates),1 as non-linear optical devices (niobium calcium borophosphates)2 and as glass seals and low-melting glass solders (zinc and calcium borophosphates).3 Recently, borophosphate glasses have received increasing attention for their use in biomedical applications due to their bioresorbable properties.4–7 They react with and dissolve in the physiological environment being eventually replaced by regenerated hard or soft tissue. Therefore, they may be used as degradable temporary implants, e.g. when such an implant is needed only for a certain length of time in order to promote healing or the growth of the surrounding tissue and in order to avoid the need for secondary surgery to remove the implant. They may also find application as drug delivery systems (antibiotics, biological active agents) as they could deliver the drugs within the body in a controlled way.8 Polymers and copolymers based on polyglycolic and polyactic acids are currently used as bioresorbable materials as they can degrade by hydrolysis. However, as they are not entirely amorphous, small crystalline particles can be released during the degradation process causing inflammatory reactions. Borophosphate glasses appear to be ideal bioresorbable materials as they do not release crystalline fragments during dissolution. However, phosphate glasses are generally hygroscopic, often highly so, and have poor mechanical properties and this can limit their use in biomedical applications.9 It has been demonstrated that the addition of B2O3 to a phosphate network improves the chemical durability as well as thermal and mechanical stability of the pure phosphate glass.10,11 Recently, it has also been shown that borate glasses have good bioactivity characteristics and thus also have much potential for use in the regeneration of tissues.12 Moreover, like the phosphate glasses, they are soluble in water; indeed, the solubility of the phosphate network can be tailored via the addition of B2O3.Borophosphate glasses are usually obtained by a conventional melt-quenching technique. However, in the past 20 years, the innovative sol–gel synthesis route for glasses has been widely investigated as an alternative to melt-quenching due to the lower energy cost and often higher purity and better homogeneity of the final products. In particular, the sol–gel method appears to be intrinsically more suitable than melt-quenching for the synthesis of systems containing volatile components such as B2O3 and P2O5 where it is therefore desirable to operate at relatively low temperatures. Furthermore, a low synthesis temperature is important for glasses designed to be used as drug delivery systems as it may allow bioactive molecules such as antibiotics and other biologically active agents to be incorporated in the glass. In the sol–gel synthesis the precursors are mixed in solution leading to glasses with a more easily controlled morphology and composition. Therefore, the dissolution rate of the sol–gel glasses, which depends on morphology and composition, can be easily controlled. However, whilst the sol–gel synthesis of silicates glasses has been extensively studied, much fundamental work remains to be done on the sol–gel synthesis of phosphates, borates and in particular on the mixed borophosphate glasses.
The sol–gel synthesis of pure phosphates glasses having P2O5 as a main component is not straightforward.13 It is not surprising therefore that the few papers published hitherto related to the sol–gel synthesis of phosphate-containing glasses relate mainly to systems having silica as the predominant component.14–17
In particular, very limited exploration has been undertaken on the sol–gel synthesis of phosphate glasses containing B2O3. Recently, a report on a sol–gel synthesis of borophosphate glasses where B2O3 is the main component (P2O5 content 3–9 mol%) has been presented.18 However, to our knowledge, there are no successful reports of sol–gel borophosphate glasses which are silica-free or alumina-free and which have P2O5 as their main component. The work reported on sol–gel lithium borophosphates systems, with a P2O5 content of up to 50 mol%, reported crystallisation of the gels after heating to only 120 °C.19
We present herein a novel sol–gel synthesis of a series of ternary glasses in the system 40(P2O5)–x(B2O3)–(60 − x)(Na2O) where the P2O5 content was kept constant at 40 mol% and x was varied between 10 and 25 mol%. Glasses with x = 20 and x = 25 mol% retain their amorphous structure up to 400 °C. The composition was chosen on the basis of previous studies on melt-quenched phosphate-based glasses used for biomedical applications where the P2O5 content was in the range 40–55 mol%.20–22 Alkyl phosphates OP(OR)x(OH)3![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) −x (x = 1,2) were used as phosphorus precursors following our recently developed synthetic route for sol–gel phosphate-based glasses.20–22 To our knowledge, this is the first time that (P2O5)–(B2O3)–(Na2O) amorphous systems have been successfully synthesised via the sol–gel method.
−x (x = 1,2) were used as phosphorus precursors following our recently developed synthetic route for sol–gel phosphate-based glasses.20–22 To our knowledge, this is the first time that (P2O5)–(B2O3)–(Na2O) amorphous systems have been successfully synthesised via the sol–gel method.
In order to understand the effect of composition upon properties, an understanding of the structure of borophosphate glasses is fundamental. However, to perform a structural characterisation of borophosphate systems is a difficult task due to the structural complexity of the network system. Therefore, structural characterisation was performed through a combination of experimental data from X-ray diffraction, thermal analysis, infrared spectroscopy, and 11B and 31P solid state NMR. Only by combining the results of complementary techniques has it been possible to derive some views on the details of the structure.
2. Experimental
2.1 Synthesis
The following chemicals were used for the synthesis of sol–gel phosphates: phosphorus pentoxide (P2O5, Aldrich, 99.99 + %), boric acid (H3BO3, Aldrich, 99.5%), sodium methoxide (Na(CH3O), Aldrich, 95%), anhydrous methanol (CH3OH, Aldrich, water < 0.01%). Due to the hygroscopic nature of the reactants, all the reaction processes were carried out in a dry box or in a nitrogen atmosphere in order to maintain anhydrous conditions. All the glassware was dried overnight at 200 °C in order to remove surface water. Fig. 1 shows a schematic of the procedures followed to synthesise borophosphate glasses.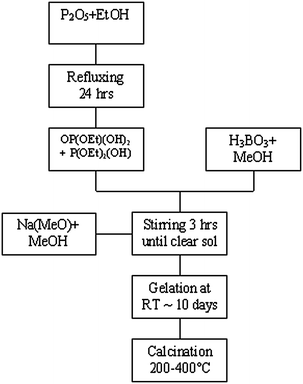 | ||
| Fig. 1 Flow diagram showing the procedure followed for synthesising borophosphate gels. | ||
The phosphorus precursor was prepared by dissolving phosphorus pentoxide (P2O5) in anhydrous ethanol:
| P2O5 + 3EtOH → OP(OEt)(OH)2 + OP(OEt)2(OH) | (1) |
2.2 Characterisation
X-Ray diffraction (XRD) patterns were obtained on a Bruker D8 Advance with Cu Kα radiation (λ = 1.54 Å), operating at 40 kV and 40 mA. Scans were performed with a detector step size of 0.02° over an angular range 2θ = 10–50° and counting for 70s per step.Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) were carried out using a Netzsch STA 409 PC at a heating rate of 5 °C min−1.
Infrared spectra were collected using a Bio-Rad FTS 175C FT-IR spectrometer controlled by Win-IR software. Samples were diluted (1:10 by weight) in dry KBr and scanned in the range 670–1500 cm−1. Each spectrum was the result of summing 64 scans.
The 31P NMR spectra were recorded on a CMX Infinity spectrometer attached to an 8.45 T magnet giving a 31P Larmor frequency of 145.77 MHz. Samples were placed in the magnet using a Doty 4 mm MAS probe and spun at ∼12 kHz. The Spinsight software was used to run one-pulse experiments with a 2.7 µs pulse length corresponding to a π/6 tip angle with a pre-acquisition delay of 10 µs. A 20 s repetition time was used and no saturation was observed. Typically, 150 scans were accumulated to obtain a good signal/noise ratio. Spectra were referenced to the resonance of ammonium dihydrogen phosphate (NH4H2PO4) at 0.9 ppm. 11B NMR spectra were acquired on a Bruker Advance II+ spectrometer attached to a 14.1 T magnet giving an 11B Larmor frequency of 192.54 MHz. Samples were placed in the magnet using a Bruker 4 mm MAS probe and spun at 12 kHz. A 2.5 µs π/2 pulse length was measured on the solid, but in order to obtain uniform excitation over all possible sites, 0.8 µs excitation pulses were used. The probe stator is made of boron nitride and thus a broad boron signal was present for spectra acquired using one pulse experiments. In order to suppress this, an echo pulse sequence was used with excitation and refocusing pulses of 0.8 and 1.6 µs respectively. The delay between the pulses was 83.33 µs. Typically, ∼2000 scans were accumulated with a recycle delay of 1 s and no saturation was observed. Spectra were referenced to the resonance of boron phosphate (BPO4) at −3.3 ppm.
3. Results
Four compositions of sodium borophosphate glasses in the system 40(P2O5)–x(B2O3)–(60 − x)(Na2O) were prepared with x = 10, 15, 20 and 25 mol%. Hereafter, samples will be named as PwByNaz where P, B, Na denote P2O5, B2O3, Na2O and w, y, z the mol% of each oxide.3.1 XRD analysis
In Fig. 2, XRD patterns of all samples after heat treatment at 200, 300 and 400 °C are shown. P40B10Na50 is partially crystalline at 200 °C and more crystalline at 300 and 400 °C. The crystalline peaks observed correspond to the phase pentasodium triphosphate Na5P3O10.23 P40B15Na45 is amorphous at 200 °C, but at 300 °C and 400 °C shows a few crystalline peaks corresponding to the phase Na5P3O10. P40B20Na40 and P40B25Na35 are mainly amorphous up to 400 °C.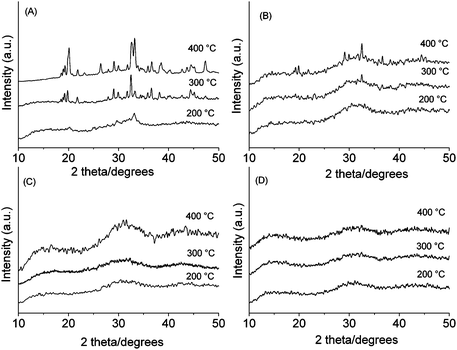 | ||
| Fig. 2 XRD patterns of: (A) P40B10Na50, (B) P40B15Na45, (C) P40B20Na40, (D) P40B25Na35. | ||
3.2 Thermogravimetric analysis (TGA) and differential thermal analysis (DTA)
TGA and DTA curves are presented in Fig. 3. The weight loss of all xerogels is complete after calcination to 270 °C. However, as reported in Table 1, xerogels with different compositions show different weight losses. The total weight loss between 25 and 270 °C is about 35% for P40B10Na50, 33% for P40B15Na45, 55% for P40B20Na40 and 62% for P40B25Na35. In P40B10Na50 and P40B15Na45, a first moderate weight loss is observed between 25 and 200 °C (about 15 wt%) and corresponds to an endothermic event as shown by the associated feature in the DTA curves. A second weight loss (about 20 wt%) is observed between 200 and 270 °C and corresponds to an exothermic event as shown by the associated feature in the DTA curve. In P40B20Na40 and P40B25Na35 a significant weight loss is observed between 25 and 150 °C (40 and 50 wt%, respectively) and a second moderate weight loss (15 and 12 wt%, respectively) is observed between 150 and 270 °C. The first weight loss is associated with a large endothermic peak, the second with a weak exothermic signal.| Sample | Weight loss% | Weight loss% | Total weight loss% | Na2O/B2O3 |
|---|---|---|---|---|
| 25–200 °C | 200–270 °C | |||
| P40B10Na50 | 15% | 20% | 35% | 5 |
| P40B15Na45 | 15% | 18% | 33% | 3 |
| 25–150 °C | 150–270 °C | |||
| P40B20Na40 | 40% | 15% | 55% | 2 |
| P40B25Na35 | 50% | 12% | 62% | 1.4 |
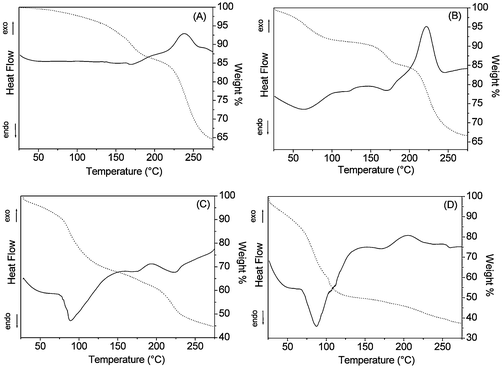 | ||
| Fig. 3 TGA (dotted line) and DTA (continuous line) curves of: (A) P40B10Na50, (B) P40B15Na45, (C) P40B20Na40, (D) P40B25Na35. | ||
The low temperature weight loss can be related to the evaporation of residual water and methanol from the hydrolysis and condensation processes remaining in the open pores of the gels, whereas the higher temperature weight loss can be related to the burning of residual organic groups in the gels.24 Results are consistent with the higher content of volatile and lower content of organic species in the samples with low Na2O/B2O3 ratio.
3.3 Infrared spectroscopy
Infrared absorption spectra are shown in Fig. 4. In all the glasses studied, the P2O5 content is higher than the B2O3 content. Therefore, the vibrational modes observed are mainly due to the phosphate network which appears in the range 700–1500 cm−1. Three main regions can be distinguished in this range: the region between 700 and 900 cm−1 is characteristic of the vibrations of bridging P–O–P groups, the region around 900–1150 cm−1 is characteristic of terminal P–O and PO3 groups and the region between 1150–1500 cm−1 is characteristic of vibrations of non-bridging PO2 groups.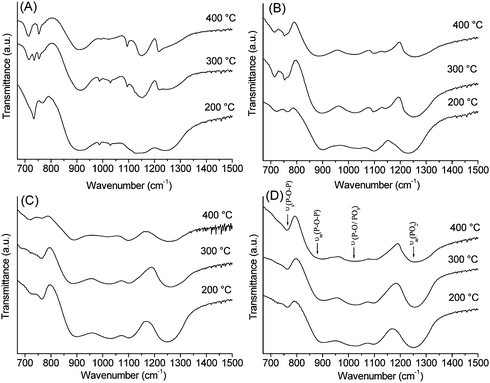 | ||
| Fig. 4 Infrared spectra in the range 670–1500 cm−1: (A) P40B10Na50, (B) P40B15Na45, (C) P40B20Na40, (D) P40B25Na35. | ||
The bands of P40B20Na40 and P40B25Na35 look similar and no significant changes are observed with the calcination temperature. Assignments of the bands are indicated in Fig. 4D. The Qn-speciation notation is used where n is the number of bridging oxygens per tetrahedron. Bands at 770 cm−1 and 900 cm−1 can be assigned to the symmetric and asymmetric stretching of the bridging oxygen atoms in a Q2 phosphate tetrahedron, ν(P–O–P), and the band at 1250 cm−1 can be assigned to asymmetric stretching modes of the two non-bridging oxygen atoms in a Q2 phosphate tetrahedron, νas(PO2).25 In addition, bands around 1000–1100 cm−1 are observed, corresponding to terminal P–O and PO3 groups (Q1 groups). Similar bands are observed in P40B10Na50 and P40B15Na45. However, in P40B10Na50 and P40B15Na45 additional sharp peaks around 700 cm−1 and in the region 900–1150 cm−1 are present, which are characteristic of crystalline phosphates.
It has to be noted that in P40B15Na45, peaks around 700 cm−1are broader then those observed in P40B10Na50 and less sharp peaks are observed in the region 900–1150 cm−1. This indicates a lower crystallinity for P40B15Na45 compared to P40B10Na50. The degree of crystallinity seems to increase with calcination temperature in both P40B10Na50 and P40B15Na45. These results are in agreement with XRD characterisation, confirming the amorphous character of P40B20Na40 and P40B25Na35, and that both P40B15Na45 and P40B10Na50 show crystallinity which is higher in P40B10Na50 and increases with calcination temperature.
3.4 MAS NMR
In order to understand the local structure and site distribution of phosphorus and boron in the samples, and their evolution with calcination temperature, 31P and 11B MAS NMR were performed. A list of possible units present in borophosphate glasses is reported in Table 2. Polyhedra around phosphorus and boron are considered separately. The phosphate units will be identified on the basis of the number of borate units connected to them, and the borate units on the basis of the number of phosphate units connected to them. The terminology Pnm will be used to define the phosphate units where n is the number of bridging oxygen atoms and corresponds to the Qn-speciation, and m is the number of next-nearest boron neighbours. Similarly, the notation Bnm will be used to define the borate units where n is the number of bridging oxygen atoms and therefore, again, corresponds to the Qn-speciation, and m is the number of next-nearest phosphorus neighbours. The 31P and 11B chemical shift ranges have been assigned on the basis of previous 11B–31P double-resonance NMR studies on borophosphate glasses which give detailed information on the boron–phosphorus interaction within the network.26,27| 31P | ||||
|---|---|---|---|---|
| Q | Name | Symbol | Unit | δ Range (ppm) |
| 3 | Branching | P 31B | OP(PO)2(OB) | −25 |
| 3 | P 32B | OP(PO)(OB) 2 | −13/−15 | |
| 2 | Middle | P 20B | OP(OP)2O− | −15/−28 |
| 2 | P 21B | OP(OP)(OB)O− | −10/−18 | |
| 2 | P 22B | OP(OB)2O− | −2/−9 | |
| 1 | End | P 10B | OP(OP)O22− | 6/−5 |
| 11B | ||||
|---|---|---|---|---|
| Q | Name | Symbol | Unit | δ Range (ppm) |
| 4 | Branching | B 43P | B(OP)3(OB) | −2/−3 |
| 4 | B 42P | B(OP)2(OB)2 | −1/−0.4 | |
| 4 | B 41P | B(OP)(OB)3 | 0.3/−0.4 | |
| Sample | Crystalline component | P 10B | P 21B | P 31B/P32B | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δ (ppm) | W (ppm) | % I | δ (ppm) | W (ppm) | % I | δ (ppm) | W (ppm) | % I | δ (ppm) | W (ppm) | % I | |
| P40B10Na50 | ||||||||||||
| 200 °C | 2.5±0.2 |
2.3±0.3 |
28±5 |
−7.6±0.4 |
9.4±0.8 |
42±5 |
−16.8±0.4 |
8.6±0.8 |
22±5 |
|||
| −4.6±0.4 |
4.1±0.8 |
7±5 |
||||||||||
| 300 °C | 2.1±0.2 |
2.1±0.3 |
23±5 |
−11.0±0.4 |
12.5±0.8 |
62±5 |
−19.6±0.4 |
6.2±0.8 |
13±5 |
|||
| −7.2±0.4 |
1.6±0.3 |
2±5 |
||||||||||
| 400 °C | 1.5±0.2 |
1.3±0.3 |
20±5 |
−10.8±0.4 |
6.7±0.8 |
40±5 |
−18.0±0.4 |
7.7±0.8 |
28±5 |
|||
| −7.2±0.4 |
1.6±0.3 |
13±5 |
||||||||||
| P40B15Na45 | ||||||||||||
| 200 °C | 1.2±0.2 |
5.7±0.7 |
7±2 |
−7.7±0.2 |
12.0±0.5 |
84±2 |
−18.4±0.2 |
6.8±0.5 |
9±2 |
|||
| 300 °C | 1.4±0.2 |
1.4±0.3 |
4±4 |
−10.3±0.2 |
10.1±0.8 |
74±5 |
−19.1±0.2 |
6.5±0.8 |
22±5 |
|||
| 400 °C | 1.4±0.2 |
2.2±0.8 |
4±4 |
−10.3±0.2 |
7.6±0.8 |
52±5 |
−17.2±0.4 |
8.6±0.8 |
44±5 |
|||
| P40B20Na40 | ||||||||||||
| 200 °C | 0.0±0.2 |
7.6±0.5 |
5±2 |
−9.3±0.2 |
9.9±0.5 |
70±2 |
−18.6±0.2 |
7.6±0.5 |
24±2 |
|||
| 300 °C | 0.3±0.2 |
7.4±0.5 |
3±2 |
−10.4±0.2 |
9.7±0.5 |
61±2 |
−19.0±0.2 |
7.8±0.5 |
36±2 |
|||
| 400 °C | 0.8±0.2 |
5.2±0.6 |
2±2 |
−10.6±0.2 |
8.5±0.5 |
52±2 |
−18.7±0.2 |
8.1±0.5 |
45±2 |
|||
| P40B25Na35 | ||||||||||||
| 200 °C | 0.3±0.2 |
8.3±0.8 |
2±2 |
−9.3±0.3 |
9.9±0.5 |
65±2 |
−18.5±0.2 |
8.7±0.5 |
33±2 |
|||
| 300 °C | 0.2±0.2 |
7.4±0.8 |
1±1 |
−10.9±0.2 |
9.7±0.5 |
54±3 |
−19.3±0.2 |
9.0±0.5 |
45±2 |
|||
| 400 °C | 0.3±0.2 |
9.0±0.8 |
2±2 |
−10.0±0.6 |
9.0±0.8 |
53±11 |
−18.5±0.2 |
9.3±0.7 |
45±14 |
|||
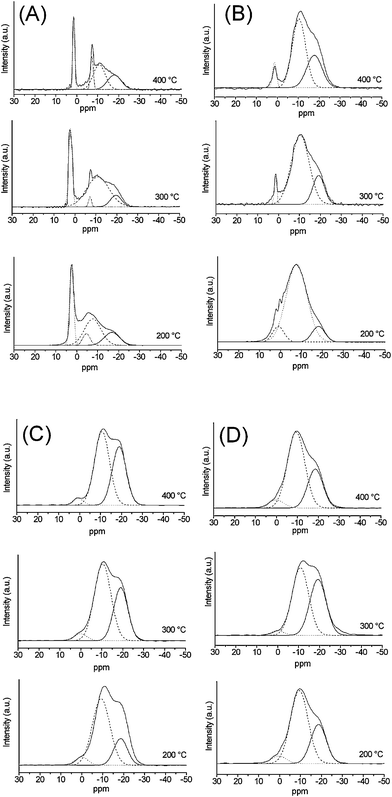 | ||
| Fig. 5 31P MAS NMR spectra from: (A) P40B10Na50, (B) P40B15Na45, (C) P40B20Na40, (D) P40B25Na35. | ||
Regardless of the calcination temperature, P40B10Na50 shows sharp peaks which indicate partial crystallisation of the samples, in agreement with XRD patterns. In particular, the samples calcined at 300 and 400 °C show two sharp peaks, the first in the range 1.5/2.1 ppm and the second at −7.2 ppm. XRD analysis shows that these samples contain well crystallised pentasodium triphosphate Na5P3O10. Therefore, the two resonances can be ascribed to two different phosphorus environments in Na5P3O10. This is confirmed by the chemical shifts values found in the literature for the two inequivalent sites Na5P3O10 at 1.3 and −7.5 ppm.28 In P40B10Na50 calcined at 200 °C, two peaks are observed at 2.5 and −4.6 ppm, which are broader compared to samples calcined at higher temperatures. This indicates a more disordered environment around the two P sites for the sample calcined at 200 °C, in agreement with the more amorphous character of the XRD pattern. In addition to the sharp peaks due to the crystalline phases, all P40B10Na50 samples show two more resonances in the ranges −8/−11 ppm and −17/−20 ppm that can be assigned to P21B groups and P31B/P32B groups, respectively.
In P40B15Na45, even after calcination, only one sharp resonance due to partial crystallinity can be observed at 1.4 ppm. The peak width decreases with increasing calcination temperature, in agreement with the higher degree of order observed with increasing heat treatment. Two more resonances corresponding to P21B groups and P31B/P32B groups are observed at all calcination temperatures in the range −8/−10 ppm and −17/−20 ppm, respectively.
In P40B20Na40 and P40B25Na35 no sharp resonances are observed, even after calcination at 400 °C, confirming the amorphous character of these compositions. At all calcination temperatures, resonances corresponding to P21B groups and P31B/P32B groups are observed in the range −9/−10 ppm and −18/−19 ppm. In addition, a very weak resonance at ∼0 ppm is observed that can be assigned to P10B species, i.e. to terminal phosphate Q1 groups. It has to be noted that in the completely amorphous systems P40B20Na40 and P40B25Na35, the relative intensities of the different speciation groups change significantly with calcination temperature, indicating significant structural changes. In particular, with the increase of calcination temperature, the relative amount of P21B decreases and the relative amount of P31B/P32Bg roups increases, indicating an increase in the connectivity of the glass with increasing temperature. However, the relative intensity of different groups does not change significantly with B2O3 content.
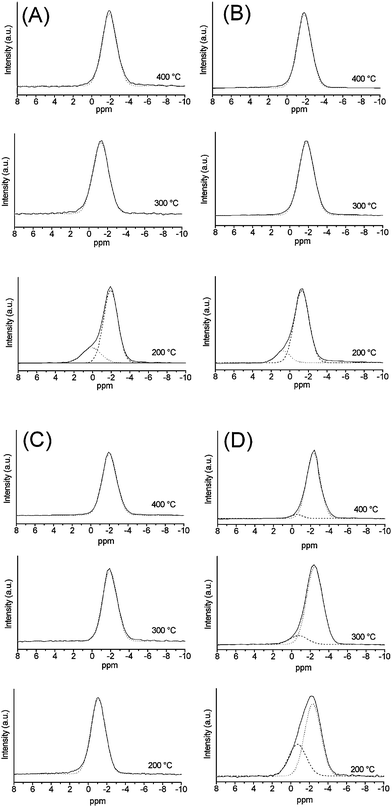 | ||
| Fig. 6 11B MAS NMR spectra from: (A) P40B10Na50, (B) P40B15Na45, (C) P40B20Na40, (D) P40B25Na35. | ||
| Sample | Q3 | B 41P | B 42P | B 43P | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δ (ppm) | W (ppm) | % I | δ (ppm) | W (ppm) | % I | δ (ppm) | W (ppm) | % I | δ (ppm) | W (ppm) | % I | |
| P40B10Na50 | ||||||||||||
| 200 °C | 15.5±2.0 |
5.5±2.0 |
5±2 |
0.1±0.2 |
2.4±0.4 |
21±5 |
−1.9±0.2 |
1.8±0.4 |
74±5 |
|||
| 300 °C | −1.3±0.2 |
2.0±0.4 |
100±1 |
|||||||||
| 400 °C | −1.9±0.2 |
1.9±0.4 |
100±1 |
|||||||||
| P40B15Na45 | ||||||||||||
| 200 °C | 0.5±0.2 |
1.7±0.4 |
13±5 |
−1.3±0.2 |
1.9±0.4 |
87±5 |
||||||
| 300 °C | −1.8±0.2 |
2.0±0.4 |
100±1 |
|||||||||
| 400 °C | −1.9±0.2 |
1.8±0.4 |
100±1 |
|||||||||
| P40B20Na40 | ||||||||||||
| 200 °C | −1.1±0.2 |
1.9±0.4 |
100±1 |
|||||||||
| 300 °C | −1.9±0.2 |
1.9±0.4 |
100±1 |
|||||||||
| 400 °C | −2.0±0.2 |
1.9±0.4 |
100±1 |
|||||||||
| P40B25Na35 | ||||||||||||
| 200 °C | −0.7±0.2 |
2.2±0.4 |
33±5 |
−2.4±0.2 |
2.0±0.4 |
67±5 |
||||||
| 300 °C | −0.7±0.2 |
2.3±0.4 |
11±5 |
−2.5±0.2 |
2.0±0.4 |
89±5 |
||||||
| 400 °C | −0.6±0.2 |
1.4±0.4 |
5±5 |
−2.3±0.2 |
1.7±0.4 |
95±5 |
||||||
The spectrum of P40B10Na50 calcined at 200 °C has been deconvoluted using two resonances at 0.1 and −1.9 ppm with intensities of 21 and 74%, respectively. Only one resonance at −1.3 ppm and −1.9 ppm is observed in the samples calcined at 300 °C and 400 °C, respectively. P40B15Na45 calcined at 200 °C has been deconvoluted using two resonances at 0.5 and −1.3 ppm with intensities of 13 and 87%, respectively. Only one resonance at −1.8 and −1.9 ppm is observed in the samples calcined at 300 °C and 400 °C, respectively. The resonance at around 0 ppm observed in the samples calcined at 200 °C can be assigned to B41P groups, the one at −1.3 ppm can be assigned to B42P and the one around −1.8/−1.9 ppm can be assigned to B43P.
In P40B20Na40 only a single resonance is observed, regardless of the calcination temperature. The resonance is observed at −1.1 ppm in the samples calcined at 200 °C (B42P groups), and at −1.9 and −2.0 ppm in the samples calcined at 300 and 400 °C (B43P groups). In P40B25Na35, peak deconvolution shows that at all calcination temperatures, the band is due to the overlap of two contributions centred at ca. −0.7 ppm (B42P groups), and −2.3/−2.5 ppm (B43P groups). The intensity of the latter resonance increases with calcination temperature, from a value of 67% at 200 °C to 95% at 400 °C.
11B NMR allows the identification of the fundamental borate coordination units and the presence of three- or four-coordinated boron.29 It has to be noted that in P40B10Na50 calcined at 200 °C, in addition to the band corresponding to four-coordinated boron, a weak resonance at 15.5 ppm, corresponding to a small amount of three-coordinated boron (5.5%) was found (not shown in Fig. 6).27
4. Discussion
The incorporation of boron into a non-siliceous sol–gel produced phosphate network is not an easy task, as evidenced, in part, by the lack of publications on the subject. The main problem with regard to obtaining silica-free amorphous sol–gel borophosphates is related to the difficulty in balancing the chemical reactivities of the boron and phosphorus precursors. Different hydrolysis reactivity of the precursors leads to inhomogeneous gels and/or crystallisation.30 Silicon alkoxides are commonly used as precursors in sol–gel synthesis of silicate glasses. However, boron and phosphorus alkoxides are extremely moisture sensitive and susceptible to rapid hydrolysis. Therefore, a modification in the choice of alkoxide precursors is necessary in order to control the hydrolysis rate of precursors in order to obtain a homogeneous gel.The main problem in the synthesis of phosphate glasses is related to the difficulty of finding a suitable phosphorus precursor. H3PO4 is too reactive towards water and usually leads to precipitation instead of gelation. On the other hand, the high stability of phosphate esters, PO(OR)3, towards water make their hydrolysis reaction very slow. Therefore, partially modified phosphorus alkoxides, where a fraction of the alkoxy groups available for hydrolysis are replaced with hydroxyl groups, were used as more convenient precursors for the sol–gel synthesis of phosphates. In the conditions used for the synthesis, the hydrolysis rate of alkyl phosphates is intermediate between H3PO4 and PO(OR)3.
Moreover, it has been reported that the relative loss of phosphorus from the gel upon heating is lower using alkyl phosphates such as OP(OR)x(OH)3−x than using P(OR)3 as a precursor.31
We have found that using a mixture of equal amounts of mono- and di-alkyl phosphates such as OP(OR)x(OH)3−x (x = 1, 2), and a fully hydroxylated boron alkoxide, H3BO3, homogeneous gels are obtained after 10 days of gelation at room temperature. The use of partially or completely modified alkoxides also reduces the amount of organic material to be removed by heating treatments. TGA curves show that the elimination of organic residues, as well as any volatile components, from gels is complete at temperatures below 270 °C.
Structural evolution of the gels with temperature depends on composition. In particular, gels having a low Na2O/B2O3 ratio (<2) evolve to fully amorphous systems which are stable up to 400 °C, whereas systems having high Na2O/B2O3 ratio (>3) evolve to mainly crystalline systems at 400 °C.
Infrared spectroscopy primarily affords information on the phosphate connectivity, due to the low concentrations of B2O3. The presence of crystalline phases in samples having a high Na2O/B2O3 ratio (>3) is confirmed by sharp vibrations typical of crystalline phosphates. The amorphous structure of samples with a low Na2O/B2O3 ratio (<2) is supported by the absence of sharp features. IR bands suggest that they are mainly formed by chains of phosphate tetrahedra connected through bridging oxygens with two other phosphate tetrahedra; the out-of-chain oxygens form terminal PO groups. However, no direct information on the interconnection of borate and phosphate units can be obtained from IR spectroscopy because of the overlap of borate and phosphate bands.
In order to reveal details of the way phosphate and borate units are linked, 31P and 11B MAS NMR analysis was used. The structural interpretation of the 11B and 31P MAS NMR is not straightforward. This is because the NMR lines are wide and not well resolved. However, information on the structures can be obtained by deconvoluting the peaks into separated resonances and by analysing the associated peak position, width and intensity of those Gaussian components.
31P MAS NMR confirms the results derived from XRD analysis. In samples with high Na2O/B2O3 ratio (>3) (x = 10, 15 mol%), sharp crystalline peaks are observed and the crystalline component decreases from x = 10 to 15 mol% B2O3. With a decrease of Na2O/B2O3 ratio (<2) (x = 20, 25 mol%), the crystallisation is suppressed at all calcination temperatures. 31P MAS NMR gives evidence of cross-linking between phosphate and borate units through one (P21B, P31B groups) or two P–O–B bonds per unit (P32B groups). P21B groups dominate the structure of all samples at all calcination temperatures. However, for a given composition, the intensity of P21B progressively decreases with the increase of temperature, whereas those of P32B, P31B increases. This observation indicates that the cross-linking of the glass increases with calcination temperature.
Information on the borate units' geometry has been obtained by 11B MAS NMR, which may be used to determine the fraction of boron in trigonal and tetrahedral coordination. Previous studies1 indicate that boron in borophosphate glasses can have trigonal or tetrahedral geometry depending on composition. 11B has a nuclear spin of 3/2 and therefore has a quadrupole moment. However the difference in the chemical shifts of the BO3 and BO4 units, which have different magnitudes of quadrupolar interactions,32 is such that at high enough applied magnetic field, second-order quadrupole broadening is sufficiently suppressed that the different peaks are well separated.33,34 The resonance associated with BO4, which has a small quadrupole coupling constant,34 is quite narrow, with a chemical shift in the range 0 to −11 ppm, whereas the BO3 unit, which has a stronger quadrupole interaction, generates a broad resonance at between 14 and 18 ppm.35 The 11B NMR data indicate that tetrahedral boron dominates at all compositions and temperatures studied. Only in P40B10Na50 is a small amount of trigonal boron present, as shown by the low intensity band at 15 ppm. 11B MAS NMR, in agreement with 31P MAS NMR results, shows evidence of cross-linking between phosphate and borate units through one (B41P groups), two (B42P groups) or three (B43P groups) P–O–B bonds per unit. For a given composition, the intensity of B43P groups increases with the temperature, becoming the main boron species for all compositions at 400 °C.
31P and 11B MAS NMR seem to indicate that the incorporation of B2O3 leads to a cross linking between the metaphosphate chains through the formation of P–O–B links. However, in the range 10 ≤ x ≤ 25, the connectivity of the samples calcined at 400 °C does not change with composition. The evidence of interconnection between phosphate and borate networks is also in accord with thermodynamical studies on phosphoborate glasses containing alkali ions,1 which indicate that P–O–B linkages are more stable relative to a mixture of P–O–P and B–O–B links.
5. Conclusion
A new sol–gel method has been developed to prepare borophosphate glasses in the system 40(P2O5)–x(B2O3)–(60 − x)(Na2O) (10 ≤ x ≤ 25 mol%). The use of a mixture of mono- and di-alkylphosphate and a methanolic solution of H3BO3 was successful in the synthesis of borophosphate gels. The elimination of organic residues as well as any volatiles from gels is complete at T < 270 °C. Systems with x = 20 and x = 25 maintain their amorphous structure after calcination even up to 400 °C, whereas systems with x = 10 and x = 15 crystallise as Na5P3O10. The crystalline character decreases with the decrease of the Na2O/B2O3 ratio. 31P and 11B MAS NMR indicate that the incorporation of B2O3 leads to a cross linking between the phosphate chains through the formation of P–O–B links.Acknowledgements
EPSRC are thanked for supporting the collaboration on bioactive phosphate glasses between The University of Kent, The Eastman Dental Institute (University College London) and the University of Warwick through projects EP/C000714/1, GR/T21080/1 and EP/C000633/1. EPSRC and the University of Warwick are thanked for their joint support of NMR facilities at Warwick.References
- A. P. Ahoussou, J. Rogez and A. Kone, J. Non-Cryst. Solids, 2007, 353, 271 CrossRef CAS
.
- V. Nazabal, E. Fargin, C. Labrugère and G. Le Flem, J. Non-Cryst. Solids, 2000, 270, 223 CrossRef CAS
- L. Koudelka, P. Mošner, M. Zeyer and C. Jäger, J. Non-Cryst. Solids, 2003, 326&327, 72 CrossRef
- A. Saranti, I. Koutselas and M. A. Karakassides, J. Non-Cryst. Solids, 2006, 352, 390 CrossRef CAS
- J. C. Knowles, J. Mater. Chem., 2003, 13, 2395 RSC
- E. A. Abou Neel and J. C. Knowles, J. Mater. Sci. - Mater. Med., 2008, 19, 377 CrossRef CAS
- I. Ahmed, E. A. Abou Neel, S. Valappil, S. N. Nazhat, D. M. Pickup, D. Carta, D. L. Carroll, R. J. Newport, M. E. Smith and J. C. Knowles, J. Mater. Sci., 2007, 42(23), 9827 CrossRef CAS
- I. Ahmed, D. Ready, M. Wilson and J. C. Knowles, J. Biomed. Mater. Res. PART A, 2006, 79A(3), 618 CrossRef CAS
- I. Ahmed, M. Lewis, I. Olsen and J. C. Knowles, Biomaterials, 2004, 25, 491 CrossRef CAS
- H. Takebe, T. Harada and M. Kuwabara, J. Non-Cryst. Solids, 2006, 352, 709 CrossRef CAS
- J. F. Ducel and J. J. Videau, Mater. Lett., 1992, 12, 271 CrossRef
- W. Liang, C. Rüssel, D. E. Day and G. Völksch, J. Mater. Res., 2006, 21, 125 CrossRef CAS
- J. Livage, P. Barboux, M. T. Vandenborre, C. Schmutz and F. Taulelle, J. Non-Cryst. Solids, 1992, 147&148, 18 CAS
- N. J. Clayden, S. Esposito, P. Pernice and A. Aronne, J. Mater. Chem., 2001, 11, 936 RSC
- R. Hsu, J. Y. Kim, P. Kumta and T. P. Feist, Chem. Mater., 1996, 8, 107 CrossRef CAS
- T. Woignier, J. Phalippou and J. Zarzycki, J. Non-Cryst. Solids, 1984, 63, 117 CrossRef CAS
- J. Y. Kim and P. N. Kumta, J. Phys. Chem. B, 1998, 102, 5744 CrossRef CAS
- M. Bengisu, E. Yilmaz, H. Farzad and S. T. Reis, J. Sol-Gel Sci. Technol., 2008, 45, 3
- A. F. Ali, P. Mustarelli, E. Quartarone, C. Tomasi, P. Baldini and A. Magistris, J. Mater. Res., 1999, 14(4), 1510 CrossRef CAS
- D. Carta, D. M. Pickup, J. C. Knowles, I. Ahmed, M. E. Smith and R. J. Newport, J. Non-Cryst. Solids, 2007, 353, 1759 CrossRef CAS
- D. Carta, J. C. Knowles, M. E. Smith and R. J. Newport, J. Non-Cryst. Solids, 2007, 353, 1141 CrossRef CAS
- D. Carta, D. M. Pickup, J. C. Knowles, M. E. Smith and R. J. Newport, J. Mater. Chem., 2005, 15, 2134 RSC
- ICSD 010-0179.
- A. Aronne, M. Turco, G. Bagnasco, P. Pernice, M. Di Serio, N. J. Clayden, E. Marenna and E. Fanelli, Chem. Mater., 2005, 17, 2081 CrossRef CAS
- J.-O. Byun, B.-H. Kim, K.-S. Hong, H.-J. Jung, S.-W. Lee and A. A. Izyneev, J. Non-Cryst. Solids, 1995, 190, 288 CrossRef CAS
- S. Elbers, W. Strojek, L. Koudelka and H. Eckert, Solid State Nucl. Magn. Reson., 2005, 27, 65 CrossRef CAS
- M. Zeyer-Düsterer, L. Montagne, G. Palavit and C. Jäger, Solid State Nucl. Magn. Reson, 2005, 27, 50 CrossRef
- C. A. Fyfe, H. M. Altenschildesche and J. Skibsted, Inorg. Chem., 1999, 38, 84 CrossRef CAS
- Y. H. Yun and P. J. Bray, J. Non-Cryst. Solids, 1978, 30, 45 CrossRef CAS
- P. N. Kumta and M. A. Sriram, J. Mater. Sci., 1993, 28, 1097 CrossRef CAS
- B. I. Lee, W. D. Samuels, L. Q. Wang and G. J. Exarhos, J. Mater. Res., 1996, 11, 134 CrossRef CAS
- N. H. Ray, Phys. Chem. Glasses, 1975, 16, 75 CAS
- M. E. Smith and E. R. H. Van Eck, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 159 CrossRef CAS
-
K. J. D. MacKenzie, M. E. Smith, Multinuclear Solid State NMR of Inorganic Materials, Pergamon, Oxford, 2002 Search PubMed
- Z. Szalay and J. Rohonczy, J. Non-Cryst. Solids, 2007, 353, 295 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2009 |