Direct electrochemistry of bilirubin oxidase on three-dimensional gold nanoparticle electrodes and its application in a biofuel cell†
Kenichi
Murata
,
Kazuki
Kajiya
,
Nobuhumi
Nakamura
* and
Hiroyuki
Ohno
Department of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, 2-24-16 Nakacho, Koganei, Tokyo, 184-8588, Japan. E-mail: nobu1@cc.tuat.ac.jp; Fax: +81 42 388 7482; Tel: +81 42 388 7482
First published on 7th September 2009
Abstract
We examined the direct electron transfer (DET) reaction of bilirubin oxidase (BOD) at three-dimensional gold nanoparticle (AuNP) electrodes in DET-type biofuel cells. The BOD-modified AuNP electrode, which does not have a thiol self-assembled monolayer, provided a current density as high as 5.2 mA cm−2 at a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm electrode rotation rate. A remarkable improvement in the stability of catalytic currents was also achieved by utilizing AuNP electrodes. Current densities retained about 90% of their initial value after 48 h of continuous measurement. Moreover, we constructed a mediator-free and compartmentless fructose/O2 biofuel cell based on DET-type bioelectrocatalysis via the BOD-cathode and the fructose dehydrogenase (FDH)-anode at pH 6.0. When carbon paper was used as the substrate upon which the electrode of the biofuel cell was constructed, the maximum current density of 2.6 mA cm−2 and the maximum power density of 0.66 mW cm−2 were achieved at 360 mV of the cell voltage in quiescent solution. Under stirring, a maximum current density of 4.9 mA cm−2 and a maximum power density of 0.87 mW cm−2 at an operating voltage of 300 mV were achieved.
000 rpm electrode rotation rate. A remarkable improvement in the stability of catalytic currents was also achieved by utilizing AuNP electrodes. Current densities retained about 90% of their initial value after 48 h of continuous measurement. Moreover, we constructed a mediator-free and compartmentless fructose/O2 biofuel cell based on DET-type bioelectrocatalysis via the BOD-cathode and the fructose dehydrogenase (FDH)-anode at pH 6.0. When carbon paper was used as the substrate upon which the electrode of the biofuel cell was constructed, the maximum current density of 2.6 mA cm−2 and the maximum power density of 0.66 mW cm−2 were achieved at 360 mV of the cell voltage in quiescent solution. Under stirring, a maximum current density of 4.9 mA cm−2 and a maximum power density of 0.87 mW cm−2 at an operating voltage of 300 mV were achieved.
Broader contextEnzymatic biofuel cells are energy conversion devices that use isolated enzymes to convert the chemical energy of a fuel into electrical energy as alternatives to the platinum catalyst in traditional fuel cells. The excellent properties of enzymes offer some interesting advantages in fuel cell applications; enzymes can oxidize various biologically-relevant organic molecules such as alcohols, sugars, and organic acids under mild conditions, typically at ambient temperature and pressure. One of the disadvantages of enzymes as electrocatalysts is their large size, which leads to a low density of the catalytic site at an electrode surface. In this study, we fabricated three-dimensional gold nanoparticle-modified electrodes and examined the direct electrochemistry of bilirubin oxidase as a cathodic enzyme and fructose dehydrogenase as an anodic enzyme at the electrodes. The gold nanoparticle assembly had a large surface area-to-volume ratio, adequate for high enzyme loading and conferring the high power output of a fructose/dioxygen biofuel cell. |
1 Introduction
Biofuel cells can generate electrical energy via the coupling of the enzymatic oxidation of biomass products (alcohols, sugars, and organic acid) with the enzymatic reduction of O2 under mild conditions (ambient temperature, near neutral pH, and ambient pressure).1–3 The cells do not require separators because cross-reactions do not occur due to the high substrate specificity of enzymes; as such, the miniaturization of biofuel cells is possible. Due to these special characteristics, biofuel cells have the potential to be used as mobile power sources.Multicopper oxidases, such as laccase, bilirubin oxidase (BOD), and CueO, are enzymes used as O2-reducing cathode catalysts.1,2BOD is widely used in the study of biofuel cells because it can catalyze the reduction of O2 at neutral pH with relatively low overpotential.22,2′-Azinobis(3-ethylbenzothiazoline-6-sulfonate) was initially reported as a mediator for BOD with a low overpotential for O2-reduction.4 Heller and coworkers later reported a set of Os complex-based redox polymers that were designed for both mediation of fast electron transfer and immobilization of the enzyme.5 The current density of O2reduction by immobilized BOD with Os-polymer was predominantly limited by the diffusion of dissolved O2. It reached about 9 mA cm−2 in an O2-saturated physiological solution (pH 7.4) with an electrode rotation rate of 4000 rpm.5 On the other hand, direct electron transfer (DET) reactions, in which enzymes directly exchange electrons with electrodes without a mediator, can be performed using many enzymes. Because mediators cause thermodynamic loss and are often harmful compounds, mediator-free DET-type biofuel cells are generally advantageous. DET-type bioelectrocatalysis by BOD has been reported for several electrodes, including carbon-based,6–15gold-based,16–19 and platinum-based20 materials. However, the catalytic currents were often only a few hundred μA cm−2 or far less. To overcome this difficulty, carbon nanomaterials have been utilized. Kano and co-workers reported that carbon nanomaterials such as a carbon aerogel are suitable materials for the DET reaction of multicopper oxidases.12 The current density based on the DET reaction of BOD at the carbon aerogel-modified glassy carbon electrode reached as high as approximately 6 mA cm−2 in O2-saturated buffer solution (pH 7.0) at 4000 rpm.
Gold nanoparticles (AuNPs) have also been utilized for the DET reaction of redox proteins such as horseradish peroxidase21 and cytochrome c22 due to biocompatibility and a large surface area-to-volume ratio. The ease of chemical modification of the surface of gold yields additional advantages for a DET reaction. Self-assembled monolayers (SAMs) of ω-functional alkanethiols are often used as stable modifiers to introduce functional groups and to control surface properties like hydrophilicity and charge.23 Taking advantage of the facility of gold surface modification, Willner and co-workers demonstrated the DET reaction of a reconstructed glucose oxidase24 and dehydrogenase25 at each enzyme's cofactor-modified AuNP assembly. Despite these advantages, AuNPs have not been widely used for DET-type bioelectrocatalysis mainly due to the difficulty of constructing a three-dimensional assembly of AuNPs possessing a large effective surface area.
Recently, we reported a simple fabrication method for three-dimensional AuNP electrodes.26 The surface area could be increased to ∼300 times greater than the projected surface area. Nanostructured electrodes have the strong advantage of possessing a three-dimensional scaffold for protein immobilization, within which the proteins themselves can act as three-dimensional conducting pathways. When fructose dehydrogenase (FDH) was immobilized on the electrode surface,27 the current density for the oxidation of D-fructose reached as high as 14.3 ± 0.9 mA cm−2 in the presence of 200 mM D-fructose. In this study, we examined the DET reaction of BOD at thiol-modified and -unmodified three-dimensional AuNP electrodes. Moreover, we constructed a mediator-free and compartmentless biofuel cell based on DET-type bioelectrocatalysis by incorporating a BOD-cathode and an FDH-anode at pH 6.0. It was shown that a DET-type biofuel cell based on three-dimensional AuNP electrodes has a high power output.
2 Experimental
2.1 Enzymes and reagents
Bilirubin oxidase (BOD; EC 1.3.3.5) from Myrothecium verrucaria (Amano Enzyme Inc., Japan) and fructose dehydrogenase (FDH; EC 1.1.99.11) from Gluconobacter sp. (Toyobo Enzymes Co., Japan) were used as received. D-Fructose (Wako, Japan) was used as the substrate for FDH. ω-Functional alkanethiols, 3-mercaptopropionic acid (MPA, from Dojindo, Japan), 2-mercaptoethanol (ME, from Kanto Chemical Co., Japan), 2-aminoethanthiol (AET, from Wako, Japan), and 1-propanethiol (PT, from Kanto Chemical Co., Japan) were purchased and used without further purification.2.2 Preparation of AuNP-modified electrodes
AuNPs were prepared following a procedure described by Frens.28 Briefly, 12.5 mL of 38.8 mM sodium citrate solution (Wako) was added to 125 mL of boiling 1.0 mM HAuCl4 (Wako) with vigorous stirring. After the appearance of a deep red color, boiling and stirring continued for 15 min. The solution was then allowed to cool to room temperature. The particle diameter of the AuNPs was estimated to be about 15 nm as calculated from the UV-vis spectrum of the solution.29 To increase the number of particles per volume, the AuNP solution was centrifuged (10000g, 30 min) in 1.5 ml Eppendorf tubes; then, 98% of the remaining supernatant volume was discarded. The precipitated AuNPs were resuspended by ultrasonication and stored as a 50-times concentrated AuNP dispersion at 4 °C. A polycrystalline gold electrode (φ = 3.0 mm, AuE) was polished with water on aluminium oxide lapping film sheets, and then 3.5 μl of the concentrated AuNPs was pipetted onto the surface of the electrode, and then the electrode was air-dried to obtain AuNP/AuE. To increase the number of particles at the electrode, the casting and evaporation steps were repeated. It has been shown that the effective surface area can be increased by increasing the number of casts with concentrated AuNPs.26 Typically, we used “three-times cast electrodes” that were prepared by repeating the casting cycle three times.
In experiments with biofuel cells, we used carbon paper (CP; SpectraCorp 2050-A) as a matrix with a highly porous three-dimensional network. In this case, the electrodes with a surface area of ∼0.25 cm2 were first anodized at +2.0 V for 15 s, followed by cathodization at −1.1 V for 15 s in 100 mM phosphate buffer (pH 7.0) to obtain a hydrophilic surface. Twenty microliters of the concentrated AuNPs were pipetted onto the hydrophilic surface of the carbon paper, and then the electrode was dried in air. This procedure was repeated six times to obtain a AuNP/CP electrode.
2.3 Preparation of enzyme-immobilized AuNP electrodes
The AuNP-modified gold electrodes were immersed in a 20 mM aqueous solution of respective thiol compounds (ME, MPA, and AET) or a 20 mM ethanol solution of 1-propanethiol (PT) for 1 h at room temperature. The resulting electrodes were thoroughly rinsed with water or ethanol to remove physically-adsorbed thiol molecules. The electrodes were then immersed in a solution containing an enzyme; for the cathode, a 1.0 mg ml−1BOD solution of 100 mM phosphate buffer solution (pH 7.0) for 2 h; for the anode, a 3.0 mg ml−1FDH solution of 100 mM acetate buffer solution (pH 5.0) for 3 h at room temperature.2.4 Electrochemical measurements
Electrochemical experiments were carried out with an ALS Electrochemical Analyzer (Model 702B). The enzyme-modified electrodes were attached to the shaft of an electrode rotator (RRDE-2, BAS) for rotating disk electrode (RDE) measurements. A platinum wire and an Ag/AgCl(3 M NaCl) electrode were used as the counter electrode and the reference electrode, respectively. Before the measurements, O2 was purged by bubbling with highly purified O2 and measurements were carried in O2-saturated buffer solution at 25 °C. All the potentials cited in this paper refer to the Ag/AgCl (3 M NaCl) electrode, with a potential of +205 mV vs. the normal hydrogen electrode (NHE).2.5 Fuel cell measurements
The obtained electrodes were positioned in a water-jacket cell containing 200 mM D-fructose solution (100 mM acetate buffer, pH 6.0) with an O2 inlet tube. A variable load (R = 10 Ω to 2 MΩ) was applied by connecting a resistor between the electrodes, and voltages (V) and currents (I) were measured via two digital multimeters (Voac 7411; Iwatsu Electric Co., Ltd, Tokyo, Japan) connected across the cell.3 Results and discussion
3.1 Cyclic voltammetry of BOD at an AuNP/AuE
Fig. 1A shows cyclic voltammograms of BOD attached to an AuNP-modified electrode without a thiol coating in an O2-saturated phosphate buffer solution of pH 7.0 at various rotation rates of the electrode. Well-defined cathodic waves were observed, and the current depended on the concentration of O2. No cathodic wave was observed at the BOD-unmodified electrode in this potential range. These results indicate that BOD electrochemically catalyzed the reduction of O2 through the DET reaction between the active center of BOD and the electrode. However, a redox response of the copper center of BOD on the AuNP electrode in the absence of O2 was not observed. The reason for this is not clear, but possible explanations include a low density of BOD per unit effective area or a broad distribution of the redox potential caused by adsorption on the electrode surface.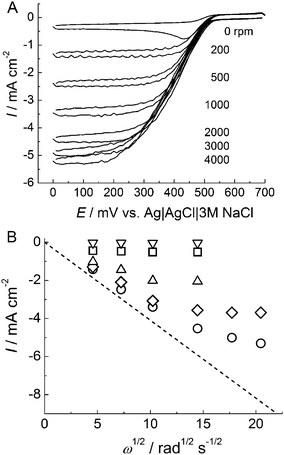 | ||
| Fig. 1 (A) Cyclic voltammograms of BOD adsorbed on AuNP/AuE under O2-saturated conditions at pH 7.0. The scan rate was 10 mV s−1 with the electrode rotation rate as indicated. (B) Levich plots for O2reduction currents at 0 V at BOD adsorbed on the thiol-unmodified AuNP electrode (circle) and thiol-modified AuNP electrodes (MPA, diamond; ME, upright triangle; AET, square; PT, inverted triangle). | ||
In a quiescent solution, a peak-shaped voltammogram was observed, suggesting the depression of O2 near the electrode surface. Typical sigmoidal-shaped voltammograms with a potential-independent plateau region were observed with rotation of the electrode. The current density at the plateau region increased with increasing rotation rate, and reached as high as 5.2 mA cm−2 at 4000 rpm (Fig. 1). The current density depended on the time of immobilization of BOD. It reached a maximum value after about 30 min (Fig. S1†). The three-dimensional nanostructured gold surface, which consists of spherical building blocks of about 50 nm, was observed by means of scanning electron microscopy (SEM).26 The effective surface area of the electrode (three-times cast electrode) was about 70 times greater than the geometric surface area of the substrate.26 This high current density was attributed to the large surface area of AuNP/AuE.
The current density was also dependent on the terminal groups of the SAMs, –COOH, –NH2, –OH, and –CH3 (Fig. 1B). When BOD was adsorbed on the carboxylate terminated SAMs-modified electrode, the electrocatalytic current of O2-reduction was higher than those on the other SAMs-modified electrodes. This agrees with the reported result from single-crystal electrode experiments.19 However, the current densities were lower than that obtained at the thiol-unmodified AuNP electrode. This result indicates that the AuNPs themselves have an excellent surface property for the DET reaction of BOD. Therefore, all following experiments were carried out using the thiol-unmodified AuNP electrode for the immobilization of BOD.
Under conditions in which O2reduction is completely limited by the mass transfer of O2, limiting currents are proportional to the square root of the angular velocity (ω1/2). The broken line in Fig. 1B represents a theoretical Levich plot for a four-electron transfer process calculated from the following parameters: concentration of O2, 1.2 mM; diffusion coefficient of O2, 1.7 × 10−5 cm2 s−1; and kinematic viscosity of water, 0.01 cm2 s−1. The deviation from the theoretical line was observed at rotation rates above 1000 rpm (ω1/2 = 10.2), suggesting that the mass transfer of O2 to the electrode surface exceeds the limitation of the O2reduction reaction at the higher rotation rates. The reductive capability of O2 at the BOD-immobilized electrode should depend on the enzyme kinetics of BOD and the amount of immobilized BOD. Mano et al. reported no deviation from the theoretical Levich plot up to 3,500 rpm (ω1/2 = 19.1) for BOD/Os-polymer assembly, indicating that BOD has a sufficient reaction rate at that rotation rate.5 Therefore, in our case, increasing the amount of redox-active BOD at the electrode surface should approach the theoretical behavior. We attempted to increase the amount of the redox-active enzyme by using electrodes with greater effective surface area. As we have reported, the effective surface area can be increased in proportion to the number of casts of the AuNPs up to at least fifteen casts. When BOD was immobilized on the electrode, however, the current density increased only slightly and reached its saturation value (ca. 5.2 mA cm−2) at a cast number of three (Fig. S2†). This phenomenon was also observed for FDH.30 On the other hand, the amount of cytochrome c could be increased linearly with the number of casts of AuNPs up to at least fifteen casts. If the mass transfer of substrate is sufficient, this difference between the proteins could be attributed to their size (cytochrome c, ∼3 nm;31FDH, ∼7 nm (the hemec-domain, 3 nm; the flavin-domain, 4 nm);32BOD, ∼6 nm in diameter19): it is presumed to be more difficult for the larger enzymes BOD and FDH to penetrate into the depths of the AuNP-assembly.
BOD has four Cu atoms that are spectroscopically and magnetically classified into three types of ions: type 1 Cu, type 2 Cu, and type 3 Cu.33 The enzyme in solution accepts electrons at the type 1 Cu site from substrates and the electrons are transferred to the trinuclear site composed of one type 2 Cu ion and two type 3 Cu ions. Finally, the electrons are donated to O2 to form water at the trinuclear site. It was shown by electrochemical experiments using site-directed mutants of the axial ligand of the type 1 Cu site that carbon electrodes can donate electrons to the type 1 Cu site of BOD.34 The catalytic waves for O2-reduction started from the formal potentials of the type 1 Cu of mutated BODs. On the other hand, other reports have suggested that the entry site of multicopper oxidases like BOD and laccase for electrons from gold electrodes is a type 2/type 3 Cu trinuclear site.17,18,35 This conclusion was derived from the fact that the onset potential of the catalytic current was very low (about +200 mV). The overpotential of O2reduction results in the loss of cell voltage of a biofuel cell. In our case, the catalytic wave began to increase at about +500 mV, as shown in Fig. 1. The potential is near the formal potential of the type 1 Cu of BOD (+460 mV).36 We conclude that type 1 Cu accepts electrons from the AuNP electrode in an analogous manner to the reaction in solution.
3.2 pH dependence of O2reduction current by BOD at AuNP/AuE
The cyclic voltammetry measurements of BOD at AuNP/AuE were performed within the pH range of 4.0–8.0. An almost constant current density of about 5 mA cm−2 was observed at a rotation rate of 4000 rpm over the broad pH range of 4.0–7.0 (Fig. 2).
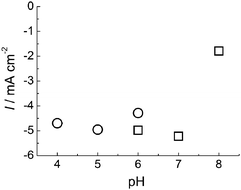 | ||
| Fig. 2 pH dependence of O2reduction current at 0 V from BOD adsorbed on AuNP electrode. Circle: 0.1 M acetate buffer. Square: 0.1 M phosphate buffer. | ||
The onset potential of reduction of O2 was dependent on pH and shifted positively with decreasing pH (−20 mV per pH unit) in a pH range of 4.0–7.0 (data not shown). This result suggests that the formal potential of type 1 Cu, which contacts the electrode and exchanges electrons directly, depends on pH. Similar pH dependences have been observed in experiments of DET reactions for BOD10,11 and CueO.37
3.3 Stability of O2reduction current by BOD at AuNP/AuE
Low stability is one of the major obstacles for the previously reported bio-devices using enzyme-modified electrodes. The stability of O2reduction current for the BOD-modified AuNP/AuE was examined in O2-saturated 0.1 M acetate buffer (pH 6.0) at 1000 rpm. An excellent stability of current was observed at the AuNP-modified electrode compared with the BOD-adsorbed polycrystalline gold electrode (Fig. 3A). The current density retained about 94% and 90% of its initial value after 24 h and 48 h, respectively. This high stability may be ascribed from the suppression of desorption of the enzyme due to the adsorption into the mesopore of the AuNP assembly. Kano and co-workers reported an improvement in the stability of direct bioelectrocatalysis by using the carbon aerogel-modified electrode with meso-type pores.12,37 The adsorption of enzyme into a mesopore is an excellent approach to improve the stability of current density.
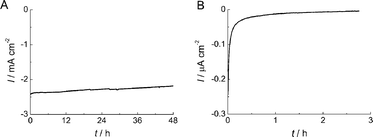 | ||
| Fig. 3 Stability of the O2reduction current for BOD adsorbed on (A) AuNP/AuE and (B) AuE under O2-saturated condition in 0.1 M acetate buffer at pH 6.0. The applied potential was +0.2 V. | ||
The stability of the FDH-adsorbed AuNP/AuE was also examined in 0.1 M acetate buffer (pH 6.0) at 1000 rpm. We have already reported that ME-SAM could be an efficient promoter of the DET reaction of FDH.27 Therefore, we also used ME-SAM to construct a FDH-anode in this study. The current density retained about 76% of its initial value after 48 h at the AuNP-modified electrode in 100 mM acetate buffer solution (pH 6.0) at a rotation rate of 1000 rpm (Fig. S3†). On the other hand, in the absence of AuNPs, the current density decreased to 13% of the initial value after 48 h. These results indicate that desorption of FDH was also successfully suppressed by using AuNPs.
3.4 Cell performance of fructose/O2 biofuel cell
The biofuel cells based on the DET-type bioelectrocatalysis for both the cathode and anode have been reported.38–42 However, the maximum power density is usually in the order of few μW cm−2 or less.38–41 Recently, Kano and co-workers reported an improvement of the power density by using carbon nanomaterials. The fructose/O2 biofuel cell based on a DET-type FDH-anode and laccase-cathode42 showed the maximum power density of 0.85 mW cm−2 at 0.41 V cell voltage under stirring condition at pH 5.0. Using nanomaterials is fundamental to construct a high performance biofuel cell.We have reported a high performance bioanode based on the DET-type bioelectrocatalysis of FDH by using three-dimensional AuNP electrodes.27 The FDH-anode showed a maximum catalytic current at pH 6.0,27 although little catalytic activity have been reported in solution and at carbon-based electrodes at the pH.32,42 This is a favourable property of the FDH-modified AuNP electrode for constructing biofuel cells at near neutral pH solution.
To construct a fructose/O2 biofuel cell, carbon paper (CP) was used as the substrate electrode. The CP has a large surface area and thus could increase the current density based on bioelectrolysis. The BOD-adsorbed AuNP/CP and FDH-adsorbed ME-AuNP/CP electrodes were combined to construct a DET-type biofuel cell without separator and mediator. Fig. 4 shows the polarization curve and the relationship between the power density and the current density of the biofuel cell in O2-saturated 100 mM acetate buffer solution at pH 6.0 containing 200 mM D-fructose. In quiescent solution, the maximum current density reached 2.6 mA cm−2 and the maximum power density of 0.66 mW cm−2 was achieved at 360 mV of the cell voltage (Fig. 4). As mentioned above, O2reduction at BOD cathode is limited by the mass transfer of O2, although oxidation of D-fructose at FDH anode is enzyme kinetics limiting in the presence of 200 mM D-fructose. These results suggest that the output of the cell is limited by the mass transfer of O2. Therefore, stirring improved the cell performance: the maximum current density reached 4.9 mA cm−2 and the maximum power density of 0.87 mW cm−2 was achieved at 300 mV of the cell voltage when stirred at 1000 rpm (Fig. 4). When the cell operated continuously for 12 h with stirring at 1000 rpm, it retained about 85% of its initial power density. The continuous high output is attributed to the robustness of the three-dimensional AuNP assembly and the stabilized enzymes in the mesopore of the electrode.
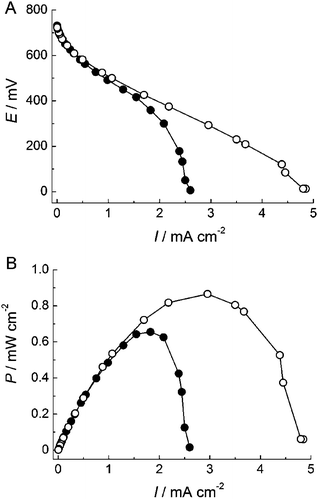 | ||
| Fig. 4 (A) Polarization curves and (B) dependence of power density (P) on current density (I) for the fructose/O2 biofuel cell. The measurements were performed in 0.1 M acetate buffer (pH 6.0) containing 200 mM D-fructose (●) without stirring and (○) with stirring solution under O2-saturated conditions at 25 °C. | ||
4 Conclusions
The fructose/O2 biofuel cell based on DET-type bioelectrocatalysis for both the cathode and the anode was constructed by using three-dimensional AuNP-modified electrodes. The gold nanoparticle electrode is preferred for the DET reaction of both cathodic enzyme BOD and anodic enzyme FDH. The electrodes showed remarkable stability of the catalytic currents. The cyclic voltammograms of BOD adsorbed on AuNP/AuE at various rotation rates of the electrodes indicated that the mass transfer of O2 is the limiting process for O2reduction. The current density for O2reduction reached as high as 5.2 mA cm−2 at an electrode rotation rate of 4000 rpm. We used CP electrodes as substrate electrodes to construct a DET-type biofuel cell. The following cell performance was achieved under stirring solution at 1000 rpm: the maximum current density reached 4.9 mA cm−2 and the maximum power density of 0.87 mW cm−2 at 300 mV of the cell voltage. The cell performance of this biofuel cell is apparently limited by a mass transfer of O2. For practical applications of biofuel cells, the necessity of stirring the solution is a critical problem. Some groups have reported an air-diffusion biocathode that can use gas phase O2.43–45 The performance of biofuel cells based on the AuNP electrode should be further improved by applying the idea of the air-diffusion biocathode.
Acknowledgements
K.M. acknowledges the financial support of the Japan Society for the Promotion of Science (Research Fellowship for Young Scientists). This study was supported by Grant-in-Aids for Scientific Research from the Japan Society for the Promotion of Science (#21225007, #21605004).Notes and references
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439 CrossRef CAS.
- S. C. Barton, J. Gallaway and P. Atanassov, Chem. Rev., 2004, 104, 4867 CrossRef CAS.
- I. Willner, Y.-M. Yan, B. Willner and R. Tel-Vered, Fuel Cells, 2009, 9, 7 CrossRef CAS.
- S. Tsujimura, H. Tatsumi, J. Ogawa, S. Shimizu, K. Kano and T. Ikeda, J. Electroanal. Chem., 2001, 496, 69 CrossRef CAS.
- N. Mano, H.-H. Kim, Y. Zhang and A. Heller, J. Am. Chem. Soc., 2002, 124, 6480 CrossRef CAS.
- S. Tsujimura, T. Nakagawa, K. Kano and T. Ikeda, Electrochemistry, 2004, 72, 437 CAS.
- S. Shleev, A. E. Kasmi, T. Ruzgas and L. Gorton, Electrochem. Commun., 2004, 6, 934 CrossRef CAS.
- S. Tsujimura, K. Kano and T. Ikeda, J. Electroanal. Chem., 2005, 576, 113 CrossRef CAS.
- M. Tominaga, M. Otani, M. Kishikawa and I. Taniguchi, Chem. Lett., 2006, 35, 1174 CrossRef CAS.
- M. Ch. Weigel, E. Tritscher and F. Lisdat, Electrochem. Commun., 2007, 9, 689 CrossRef CAS.
- K. Otsuka, T. Sugihara, Y. Tsujino, T. Osakai and E. Tamiya, Anal. Biochem., 2007, 370, 98 CrossRef CAS.
- S. Tsujimura, Y. Kamitaka and K. Kano, Fuel Cells, 2007, 7, 463 CrossRef CAS.
- F. Gao, Y. Yan, L. Su, L. Wang and L. Mao, Electrochem. Commun., 2007, 9, 989 CrossRef CAS.
- Y.-M. Yan, O. Yehezkeli and I. Willner, Chem.–Eur. J., 2007, 13, 10168 CrossRef.
- K. Schubert, G. Goebel and F. Lisdat, Electrochim. Acta, 2009, 54, 3033 CrossRef CAS.
- Y. Kamitaka, S. Tsujimura, T. Ikeda and K. Kano, Electrochemistry, 2006, 74, 642 CAS.
- R. Dronov, D. G. Kurth, F. W. Scheller and F. Lisdat, Electroanalysis, 2007, 19, 1642 CrossRef CAS.
- P. Ramírez, N. Mano, R. Andreu, T. Ruzgas, A. Heller, L. Gorton and S. Shleev, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 1364 CrossRef CAS.
- M. Tominaga, M. Ohtani and I. Taniguchi, Phys. Chem. Chem. Phys., 2008, 10, 6928 RSC.
- Y.-M. Yan, I. Baravik, R. Tel-Vered and I. Willner, Adv. Mater., 2009 DOI:10.1002/adma.200900206.
- J. Zhao, R. W. Henkens, J. Stonehuerner, J. P. O'Daly and A. L. Crumbliss, J. Electroanal. Chem., 1992, 327, 109 CrossRef CAS.
- K. R. Brown, A. P. Fox and M. J. Natan, J. Am. Chem. Soc., 1996, 118, 1154 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877 CrossRef CAS.
- M. Zayats, E. Katz, R. Baron and I. Willner, J. Am. Chem. Soc., 2005, 127, 12400 CrossRef CAS.
- K. Murata, K. Kajiya, M. Nukaga, Y. Suga, T. Watanabe, N. Nakamura and H. Ohno, Electroanalysis Search PubMed , in press.
- K. Murata, M. Suzuki, K. Kajiya, N. Nakamura and H. Ohno, Electrochem. Commun., 2009, 11, 668 CrossRef CAS.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20 Search PubMed.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215 CrossRef CAS.
- K. Murata, N. Nakamura, and H. Ohno, unpublished work.
- R. E. Dickerson, T. Takano, D. Eisenberg, O. B. Kallai, L. Samson, A. Cooper and E. Margoliash, J. Biol. Chem., 1971, 246, 1511 CAS.
- M. Tominaga, C. Shirakihara and I. Taniguchi, J. Electroanal. Chem., 2007, 610, 1 CrossRef CAS.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563 CrossRef CAS.
- Y. Kamitaka, S. Tsujimura, K. Kataoka, T. Sakurai, T. Ikeda and K. Kano, J. Electroanal. Chem., 2007, 601, 119 CrossRef CAS.
- S. Shleev, A. Christenson, V. Serezhenkov, D. Burbaev, A. Yaropolov, L. Gorton and T. Ruzgas, Biochem. J., 2005, 385, 745 CrossRef CAS.
- S. Tsujimura, A. Kuriyama, N. Fujieda, K. Kano and T. Ikeda, Anal. Biochem., 2005, 337, 325 CrossRef CAS.
- S. Tsujimura, Y. Miura and K. Kano, Electrochim. Acta, 2008, 53, 5716 CrossRef CAS.
- V. Coman, C. Vaz-Domínguez, R. Ludwig, W. Harreither, D. Haltrich, A. L. De Lacey, T. Ruzgas, L. Gorton and S. Shleev, Phys. Chem. Chem. Phys., 2008, 10, 6093 RSC.
- A. Ramanavicius, A. Kausaite and A. Ramanaviciene, Biosens. Bioelectron., 2005, 20, 1962 CrossRef CAS.
- K. A. Vincent, J. A. Cracknell, O. Lenz, I. Zebger, B. Friedrich and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16951 CrossRef CAS.
- A. Ramanavicius, A. Kausaite and A. Ramanaviciene, Biosens. Bioelectron., 2008, 24, 761 CrossRef CAS.
- Y. Kamitaka, S. Tsujimura, N. Setoyama, T. Kajino and K. Kano, Phys. Chem. Chem. Phys., 2007, 9, 1793 RSC.
- H. Sakai, T. Nakagawa, Y. Tokita, T. Hatazawa, T. Ikeda, S. Tsujimura and K. Kano, Energy Environ. Sci., 2009, 2, 133 RSC.
- R. Kontani, S. Tsujimura and K. Kano, Bioelectrochemistry, 2009, 76, 10 CrossRef CAS.
- N. S. Hudak, J. W. Gallaway and S. C. Barton, J. Electrochem. Soc., 2009, 156, B9 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data. See DOI: 10.1039/b912915d |
| This journal is © The Royal Society of Chemistry 2009 |