Carbon nanotubes—electronic/electrochemical properties and application for nanoelectronics and photonics†
Vito
Sgobba
and
Dirk M.
Guldi
Friedrich-Alexander-Universität Erlangen-Nürnberg, Department of Chemistry and Pharmacy, Interdisciplinary Center for Molecular Materials (ICMM), 91058, Erlangen, Germany
First published on 24th October 2008
Abstract
The fundamental chemical, redox, electrochemical, photoelectrochemical, optical and optoelectronic features of carbon nanotubes are surveyed with particular emphasis on the most relevant applications as electron donor/electron acceptor or as electron conductor/hole conductor materials, in solutions and in the solid state. Methods that aim at p- and n-doping as a means to favor hole or electron injection/transport are covered as well (critical review, 208 references).
Vito Sgobba received his “Laurea” in Chemistry from the University of Bari (Italy) in 2000 under the supervision of Prof. F. Naso and his PhD in Material Engineering from the University of Lecce (Italy) in 2003 with Prof. G. Vasapollo. During his PhD studies he joined Prof. L. De Cola’s group at the University of Amsterdam for a one-year Marie Curie Fellowship. Since the beginning of 2005 he has been a postdoctoral fellow with Prof. D. M. Guldi, at the Friedrich-Alexander University in Erlangen (Germany). His research interests are: synthesis, characterization, and assembly of hybrid (organic–inorganic) nanostructured materials for solar energy conversion devices. |
Dirk M. Guldi graduated from the University of Cologne (Germany) in 1988, from where he received his PhD in 1990. In 1992, after a postdoctoral appointment at the National Institute of Standards and Technology, he took a research position at the Hahn-Meitner-Institute Berlin. After a brief stay as a Feodor–Lynen Stipend (Alexander von Humboldt Foundation) at Syracuse University he joined in 1995 the faculty of the Notre Dame Radiation Laboratory where he was promoted to Associate Scientist in 1996. In 1999 he completed his Habilitation at the University of Leipzig (Germany). Since 2004 he is Professor of Physical Chemistry at the Friedrich-Alexander University in Erlangen (Germany). He was awarded with the Heisenberg-Prize (1999; Deutsche Forschungsgemeinschaft), Grammaticakis-Neumann-Prize (2000; Swiss Society for Photochemistry and Photophysics), JSPS Fellowship (2003; The Japan Society for the Promotion of Science and JPP-Award (2004; Society of Porphyrins and Phthalocyanines). His primary research interests are in the areas of new multifunctional carbon-based nanostructures within the context of light-induced charge separation and solar-energy conversion. |
Introduction
Carbon nanotubes (CNT) offer exciting opportunities for science and applications. In recent years, CNT research has been established as a highly interdisciplinary field to exploit their outstanding features.1 Conceptually, single wall carbon nanotubes (SWCNT) are considered as small strips of graphene sheets that have been rolled up to form perfect seamless single-walled nanocylinders. The way the graphene sheets are wrapped varies largely and is represented by a pair of indices (n,m). These integers relate the structure of each SWCNT to both its diameter and chirality. In other words, each SWCNT has a distinct set of (n,m). SWCNT, where the difference between n and m is a multiple of three, are metallic—all remaining ones have semiconducting character. The diameter of most SWCNT is about 1 nm, while their length reaches into the order of centimeters. Considering the small diameter and the large aspect ratios of SWCNT, they emerge as ideal one-dimensional quantum wires.2 Given their nanoscale diameters, quantum electronic confinement determine the electronic structure of CNT. Multiwall carbon nanotubes (MWCNT) are similarly regarded as coaxial assemblies of SWCNT cylinders that are, however, placed within another. The simplest representative of a MWCNT is a double wall carbon nanotube (DWCNT).The structure of CNT is entirely composed of sp2-hybridized carbons, which are notably stronger than the sp3-hybridized carbons in diamond. As a matter of fact, CNT exhibit good chemical stability, unique tensile strength (i.e., 100 times stronger than steel and 10 times stronger than Kevlar) and outstanding elastic Young's modulus (i.e., 7 times that of steel). Moreover, CNT with a surface area up to 1500 m2 g−1 are lighter than aluminum, are thermally stable at temperatures exceeding 1000 °C, and have a thermal conductivity that is, at 6000 W mK−1 twice that of diamond.3
Importantly, electrons move—depending on the CNT arrangement—differently in the tubes, which leads to either semiconducting or metallic properties.4 The electrical transport in good quality metallic SWCNT is ballistic, that is, electrons do not suffer from any scattering event over a length scale of several micrometers and/or from any electromigration, even at room temperature. As a consequence, they may carry current densities ca. 1000 times that of a typical copper wire.5 For semiconducting SWCNT the electron transport is also ballistic, but only in dimensions of a few hundred nanometers.
All known CNT preparation methods6—laser ablation, electric arc discharge, catalytic disproportionation of gaseous hydrocarbon iron (HiPCO) or cobalt molybdenum catalyzed (CoMoCat), metal organic chemical vapor deposition (MOCVD)—lead to mixtures of CNT that exhibit different chiralities, diameters and length. In addition, non-CNT carbon and metal catalysts are present in the final material. It should be noted that the removal of these byproducts/impurities is more costly than the production itself.
Early progress in understanding the optical characteristics of SWCNT was hampered by their aggregation into insoluble bundles and/or ropes when synthesized,7 causing a mixing of the energy states of different CNT structures. In fact, the high polarizability and the smooth surface of SWCNT are the inception to strong van der Waals interactions, reaching values as high as 500 eV per 1 μm of CNT length.8 Such features are responsible for the low solubility and, in turn, the low reactivity of SWCNT in common organic solvents. However, the preparation of functional CNT based nanocomposites necessitates the disentanglement of bundles and/or ropes.9 To this end, the chemical functionalization of CNT surfaces has attracted growing interest as a potent means to debundle and/or disperse them.14 Nevertheless, debundling by ultrasonication is the most widely applied method10 despite the fact that controlled conditions are key requisites to avoid/to limit damages to the sidewalls that occur during the ultrasonication treatment.11 Recently, significant progress has been made in the fields of synthetic and post-synthetic methods toward SWCNT with well defined diameters, lengths, chiralities and electronic properties.12 During the same time span, a number of techniques have been established that allow the systematic debundling, separation and characterization of different SWCNT.13
In the context of chemical reactivity, CNT are regarded as either sterically bulky π-conjugated ligands or as electron deficient alkenes. Their shells of sp2-hybridized carbons form highly aromatic hexagonal networks that are susceptible to a wide range of chemical reactions.14 Small diameter SWCNT15 are more reactive than those that posses larger diameters.16 An important aspect, especially when considering the separation of different (n,m) SWCNT, is the fact that metallic CNT are slightly less aromatic than semiconducting ones,17 and, in turn, are more reactive than the latter.13a,c
A simple structural consideration suggests that CNT might be far more reactive at their ends than in the areas along the sidewalls. It is mainly the increased curvature that is responsible for such a conclusion.18,14c Notwithstanding, the sidewalls show upon closer inspection some fairly reactive sites—carboxylic groups and other oxygenated sites. Such functional defects are formed during the growth of CNT or introduced during post processing applications (i.e., purification, separation, etc.). Depending on the actual conditions of such post treatment the defect sites might amount to around 1–3% of all carbon atoms present.19 Topological defects are also present in CNT. Here, pentagon–heptagon pairs, which are typically referred to Stone–Wales or 7/5/5/7 defects, play an important role. These structural imperfections augment the curvature along the sidewalls.19b,20 Alternatively, the defects might originate from (i) substitutional dopant impurities, (ii) structural deformation, especially bending or twisting of CNT, and (iii) vacancies in the CNT lattice.21 Most of the physical and chemical properties of the CNT are impacted by the aforementioned defects.22 A remarkable demonstration of this effect is the rectifying behavior in some SWCNT that originates exclusively from naturally occurring defects.22b
A comprehensive understanding of CNT chemistry, electrochemistry and photophysics evolves not only as a necessity but also as a central fundament for applications in artificial photosynthesis23 and in renewable energies.24
Excited, reduced and oxidized states
Excited states
Employing absorption spectroscopy of SWCNT suspensions shed light onto their electronic states. Important is in this context that 1 nm diameter tubes have a band gap of approximately 1 eV, which is close to that of silicon. But unlike silicon, semiconducting SWCNT are direct band gap materials (the minimum energy of the conduction band lies directly above the maximum energy of the valence band in momentum space) and, as such, they are known to directly absorb and/or emit light.As already mentioned, CNT tend to aggregate when synthesized and, thereby, to limit optical studies to probe bundles of SWCNT rather than individual SWCNT. In particular, the absorption spectrum of bundles exhibits severe inhomogeneous broadening as the result of mixing energy states of different CNT structures.25 To circumvent this problem, CNT must be individualized by, for example, encasing them in micellar assemblies. A common approach encompasses the ultrasonic agitation of aqueous dispersions of SWCNT in sodium dodecyl sulfate (SDS) followed by a centrifugation step to remove bundles, ropes, and residual catalyst.10 Equally successful is the wrapping with polymers26 or DNA.27
SWCNT suspensions—as prepared by one of the aforementioned means—reveal three set of bands that all arise from electronic transitions between different van Hove singularities. For HiPCO SWCNT, bands centered in the 950–2000 nm and 700–950 nm range are ascribed to transitions that arise between the first (S11) and second (S22) singularities in semiconducting SWCNT, respectively. Metallic SWCNT, on the other hand, exhibit bands in the 400–700 nm, again resulting from transitions between first (M11) singularities. (See Scheme 1)28 Typically, the absorption spectrum is best described as superpositions of narrower absorption bands corresponding to individual SWCNT having different (n,m).29
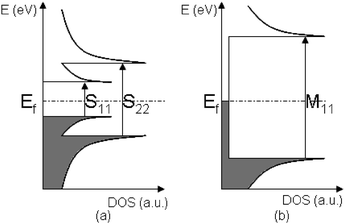 | ||
| Scheme 1 Energy diagram showing the density of states in semiconducting SWCNT—left—and metallic SWCNT—right—valence bands are grey, while conduction bands are white. | ||
Electrostatic electron/hole interaction energies (i.e., exciton binding energies) in SWCNT are significant. To be precise: on the order of 0.3 to 0.5 eV at band gap energies of ca. 1 eV.30 An immediate consequence of this strong electron/hole attraction is that a general perception treats photoexcited states of CNT excitonic (i.e., electron/hole pairs) rather than as uncoupled electrons. In SWCNT, excitons are characterized by electron/hole separations (i.e., Bohr radius) of approximately 2.5 nm.30a,31
Interestingly, fluorescence is hardly observed from SWCNT bundles. Photoexcited carriers that are generated, for example, in semiconducting SWCNT, which actually do fluoresce, relax along efficient nonradiative pathways in metallic SWCNT that are statistically present in bundles.32 Fluorescence measurements with individual SWCNT that have been isolated in SDS aqueous surfactant suspensions have instead revealed distinct electronic absorption and emission transitions for more than 30 different species.33 Inhomogeneous broadening and spectral overlap between fluorescence from different CNT structures hamper, nevertheless, recording fine resolved fluorescence spectra. The Stokes shifts are typically around <64.5 cm−1,34 and the fluorescence quantum yields are on the order of 10−4. The overall low emission efficiency has been rationalized on the basis of multiple dark excitonic states that are situated below the lowest lying bright excitonic states.35 Nevertheless, a mixture of few specific (n,m) semiconducting SWCNT were selectively suspended in toluene by using commercial poly[9,9-dioctylfluorenyl-2,7-diyl] as a dispersing and stabilizing agent.36 Here, a surprisingly high emission quantum yield of ca. 1% was measured.37 Notably, photoluminescence in the visible from MWCNT has been observed using infrared excitation suggesting efficient multi-photon absorption.38
Resonance Raman spectroscopy emerged as another powerful technique in the context of characterizing SWCNT. Notable is that allowed optical transitions between different van Hove singularities and/or excitonic transitions lead to a marked resonance enhancement of the Raman scattering. By combining data that are acquired through fluorescence and resonance Raman investigations, each optical transition has been mapped out and assigned to specific (n,m) SWCNT.33,34
Complementary work, in which the fluorescence of SWCNT that were suspended in aqueous solutions of polyacrylic acid, disclosed an excited state decay on the time scale of 10 ps. Such a fast relaxation must be connected to nonradiative processes. Taken this into consideration the low fluorescence quantum yields could be explained. From the measured decay rate and a determination of fluorescence quantum efficiency a radiative lifetime of 110 ns was deduced.39
Epifluorescence confocal microscopy performed with samples that were spun from SDS/water or dichloroethylene onto quartz enabled imaging stable emissions from single SWCNT.32 Choosing excitation at 633 nm ensured that all the Raman signals between 633 and 770 nm are distinctively different from the fluorescence signals above 850 nm. The emissive peaks have a simple narrow Lorentzian line shape and a polarization dependence, as expected for a one-dimensional system.
The local environment has a profound impact on the optical features of SWCNT. In fact, the peak positions and the line width—absorptive and emissive—from the same (n,m) SWCNT differs when dissimilar surfactants are utilized. Further alterations may arise when trap states come into play as they may evolve from structural and/or chemical defects. Moreover, the fluorescence tends to red-shift with increasing temperature40 and is also affected from mechanical strains,41 electric42 and magnetic fields.43 Notably, unlike almost all other known emitters—including small molecule and quantum dots—the single SWCNT fluorescence fails to blink at room temperature.32
Their photostability, that is, their resistance to photobleaching, together with their versatile wavelength tunability—they are optically sensitive to light in the range between 800 and 1100 nm and emit between 1300–1500 nm—render SWCNT ideal single-molecule fluorophores.44 They can also be pumped electrically to generate strong electroluminescence in the near-infrared.45
Ultrafast transient absorption spectroscopic measurements complement the single molecule studies and have been used to clarify the time scales and nature of ground state recoveries in CNT and to extract information about excitonic lifetimes. For relaxation from the excited state, an omnipresent fast decay component (300 to 500 fs) is likely due to bundled SWCNT and/or metallic SWCNT. A much slower decay component (100 to 130 ps) only appears when probing on resonance for semiconductor SWCNT and likely corresponds to the intrinsic excited state lifetime of photoexcited excitons. Lifetimes in the 100 to 130 ps range were corroborated by photoluminescence lifetime39 and by correlated single photon counting spectroscopic measurements.34
If the excitation intensity is increased, multiple electron/hole pairs are generated in SWCNT. However, these e-h pairs deactivate each in less than 3 ps. The energy released by the annihilated exciton is used to excite a second exciton to a higher energy level.
Reduced states
The electron acceptor properties of CNT and the factors that control these, need careful considerations as they impact the reactivity of the reduced state, their photochemistry and photophysics. They also tie into electron storage capabilities and may answer questions on how the electron storage changes the overall energetics. Finally, aspects on how to integrate them properly into photonic, electrochemical and electronic devices should not be neglected.Reductions of SWCNT bundles were achieved in solid films by intercalating them with alkali metals46 and/or anion radicals47 and followed spectroscopically by in situ Raman48 and vis–near-infrared measurements.46,49 In the vis-near-infrared absorption spectrum, the most profound changes are seen in the bleaching of the optical transitions associated with filling the corresponding electronic states. The gradual disappearance is noted for the S11, S22 and the M11 transitions—although the latter are impacted only to a minor extent—in all cases.47,48,50 For instance, exposure of SWCNT to fluorenone–lithium and anthraquinone–lithium attenuates exclusively the S11 peaks. On the contrary, reaction with naphthalene–lithium yields heavily reduced SWCNT, in which the M11 completely disappear. Concomitant with this disappearance is the gradual growth of new features that are positioned between those of S11 and S22. The nature of this newly evolving band is still unclear at this point.46,50,51
In resonance Raman measurements, the radial breathing mode (RBM) bands are localized in the range between 160 and 365 cm−1. Utilizing different laser energies assisted in resolving this part of the spectrum for several different (n,m) HiPCO SWCNT deposited onto conducting electrodes. This led to the experimental Kataura plot.28a,52 Consequently, RBM-bands are suitable for investigating single (n,m) SWCNT effects. At first glance, when SWCNT are reduced, the intensities of the RBM-band drop due to an inherent loss of the resonant conditions (i.e., bleaching of the S11, S22 and M11 optical transitions).53 Under very strong reductive conditions also a blue-shift of these frequencies is observed. This is ascribed to charge induced C–C bond contractions and to a charge induced SWCNT debundling.54 A similar intensity loss characterizes the G–band in the tangential vibrational modes. Again, a loss of resonance enhancement is responsible. Nevertheless, the frequency and the shape of the G–band appear to be more sensitive to reduction than the RBM-band.55 The electrons injected into the conduction bands of SWCNT should weaken the sp2 character and, in turn, red-shift the G–band as observed in other carbon based materials.56
The electronic structure can also be tuned electrochemically by controlling the interfacial potential of a SWCNT film as a working electrode. In this case the SWCNT film is immersed in an electrochemical cell and can be a pure buckypaper55,57 or cast on an inert back electrical contact (Pt, Au or Hg)53a,5,58 in contact with an electrolyte solution (0.1 M KCl) where a Pt counter-electrode and a Ag/AgCl reference electrode is provided. Implicit is that the electrochemical tuning is much easier and more precise when compared with the chemical reduction. In order to increase the electrochemical window, especially in the cathodic range (i.e., less than −0.3 V), an Hg electrode is appropriate because the disturbing evolution of H2 is minimized.
Typical cyclic voltammograms (CV) display over the entire scanning range—from, for example, −0.5 to +0.7 V—only a monotonous charge injection behavior. Double-layer charging and overlapping peaks of the charge transfer to the van Hove singularities of individual (n,m) SWCNT in the mixture lead to this feature. In protic solvents a faradaic contribution emerges between +0.2 V and −0.2 V from an involvement of carboxylic groups—see Scheme 2.58a
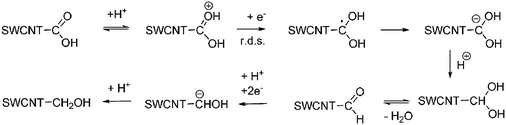 | ||
| Scheme 2 Reductive mechanism involving the carboxylic groups bound to SWCNT. | ||
In situ spectroelectrochemistry, where bulk spectroscopy and phase boundary electrochemistry are complementary combined, is, nevertheless, better suited to gather details about the redox chemistry of SWCNT. For example, individual SWCNT, which were deposited from diluted water/SDS suspensions were tested by in situ micro-Raman spectroelectrochemistry.58f,59 Aprotic electrolytes such as acetonitrile,58a,29a ethylene carbonate/dimethylcarbonate, tetrahydrofuran57a and especially ionic liquids such as fluoroborate 1-butyl-3-methylimidazolium salts allow exploring a broad window of electrochemical potentials.58e
Overall, the spectroelectrochemical results are a qualitative match of the visible/near-infrared spectra recorded upon chemical reduction of SWCNT—vide supra. Additional benefits of this method are the establishment of the reversibility of the reduction and the rate of reduction.47,51,60
Recently, the redox properties of alkali metals reduced SWCNT61 were determined in dry and deoxygenated DMSO solution containing tetrabutylammonium hexafluorophosphate as supporting electrolyte. The potentials were kept under −0.6 V to avoid additional reduction of sodium. The voltammetric and visible/near-infrared spectroelectrochemical investigations showed a 35 meV red-shift of the absorption peaks when compared with D2O/SDS suspensions and a red-shift of 15 meV when compared with metal-free DMSO suspensions.62
An accurate analysis of this data enabled calculating the average standard reduction potentials of several (n,m) SWCNT. The latter were found to be in good agreement with the experimental values that were reported for SWCNT elsewhere.63
Oxidized states
Oxidation has been among the first chemical reactions that were tested with CNT64 and is still a key step in their purification—oxidative removal of Fe, Co or Ni catalyst and of amorphous/graphitic carbon impurities.65 When opening of the hemispherical caps, shortening and chemical functionalization accompanies in many instances the oxidative treatment.As SWCNT are known to exist in bundles, which are composed of up to hundreds of individual tubes, their oxidation takes mainly place at the end tips and to a lesser amount at the defect sites. The reactivity is, nonetheless, limited to the outer layer of the bundles.66 Although the oxidation of MWCNT acts, in principle, on the same sites, it is routinely employed for MWCNT thinning through the systematic removal of the outer layers.67
O2—in its triplet ground state—does not oxidize CNT. Instead, it physisorbs quite strongly to the sidewalls of CNT. Isolated, apparently semiconducting CNT can be converted into an apparent metal through oxygen exposure at room temperature. Upon photoexcitation, this physisorbed O2 desorbs from the CNT sidewalls.68 On the other hand, the much more reactive singlet oxygen—generated, for example, by UV photoexcitation—affords either [2+2] and [4+2] cycloadducts with a preference to react with metallic SWCNT (see Scheme 3). The reaction itself is accompanied by quenching of the photoluminescence. Then, upon thermal activation, the cycloadducts readily undergo O-O bond cleavage.69 This procedure was conveniently adapted to separate CNT with different chiralities.70
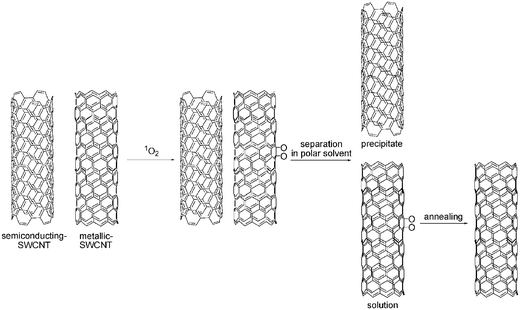 | ||
| Scheme 3 Reversible cycloaddition of singlet oxygen to SWCNT and separation of semiconducting SWCNT from metallic SWCNT. | ||
Oxidative treatment of CNT was performed under a wide variety of experimental conditions. Hereby, sonications in oxygen containing acids (i.e., HNO3, H2SO4, H2SO4 plus K2S2O8, HNO3 plus H2SO4, HNO3 plus supercritical water, trifluromethanesulfonic and chlorosulfonic acid, peroxytrifluoroacetic acid, H2SO4 plus KMnO4, H2SO4 plus H2O2, HClO4, H2SO4 plus K2Cr2O7) play the most dominant roles. As a consequence of such treatments, carboxylate groups distributed over the CNT surface are generated. In the presence of H2SO4, also sulfate ketone, phenol, alcohol and ether groups were registered. Other often utilized oxidants are: dilute ceric sulfate, H2O2, O2 plasma and RuO4.1h
Oxidation leads to the loss of structure in the ultraviolet/visible/near-infrared spectrum as a consequence of converting large numbers of sp2-hybridized carbons to sp3 analogs, which leads to altered electronic structures of CNT.14f The concentrations of carboxylic acid that are formed during the oxidative treatment were determined by evaluating the CO(g) and CO2(g) concentrations at high temperatures71 or measuring the atomic oxygen percentage with calibrated energy-dispersive X-ray spectroscopy (EDX).72 Chemical titration assays emerged as alternative tests to macroscopically estimate the defect density in CNT. Titrations of the purified SWCNT with NaOH and NaHCO3 solutions were used to determine the total percentage of acidic sites and carboxylic acid groups, respectively.73 A recent study also highlighted the opportunity to label the carboxylic groups with TiO2nanoparticles as markers.74 Interesting is the observation that a MWCNT/TiO2 composite reveals a pronounced photocatalytic activity towards the photodegradation of phenol.75
The carboxylic acid groups, introduced during the oxidation reaction, have conveniently been converted to ester and/or to amide functionalities. The chart, shown in Scheme 4, summarizes some representative cases.1h
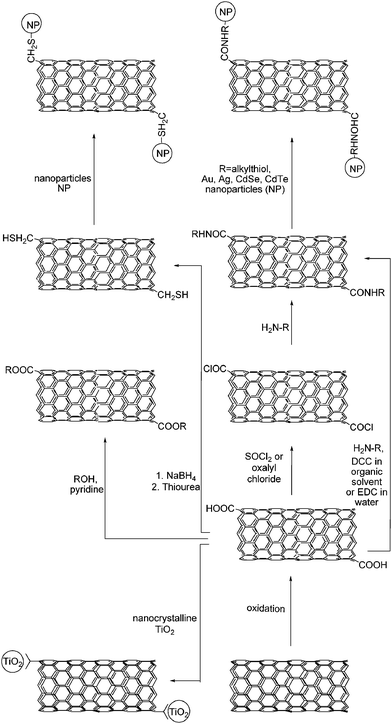 | ||
| Scheme 4 Derivatization of carboxylic functionalities present on CNT. | ||
The effect of CNT chemical and electrochemical oxidation has been extensively studied with electrochemistry, photoelectrochemistry and in situ visible/near-infrared and Raman spectroelectrochemistry. The oxidation processes are commonly accompanied by the gradual disappearing of S11, S22 and M11 bands and the growth of a new optical absorption band between S11 and S22. Raman spectroelectrochemistry shows a reversible drop of the intensities of both RBM- and G-modes.53a,58e Going beyond 1.2 V, an irreversible breakdown occurs, due to the photoanodic decomposition of SWCNT.53a,58a A strong blue-shift of the RBM is also observed between 0.5 to 1 V. The G-band blue-shifts too, because the expected stiffening of graphene is connected to the introduction of holes in the π band.76 The intensity of the disordered D-band tends to grow irreversibly upon both oxidation58b and reduction.57a
Doping
The doping of SWCNT and other graphene like species is generally amphoteric,48,46 that is, both electron or hole doping (n- or p-doping) are accessible by changing the chemical dopant or by tuning the electrochemical potential applied to the nanocarbon. The hole or the electron conductivity—together with the thermal conductivity—can be respectively raised many orders of magnitude, well above the intrinsic conductivity of the pure materials.In terms of the rigid band structures, doping of CNT simply shifts the Fermi level EF, while the band structure (DOS) remains intact. The position of EF in undoped material is assumed to be in the middle of the gap between the van Hove singularities. The absolute EF positions and the work functions of individual isolated SWCNT are not identical, but scale linearly with the gap energy.58f,59,77 The doping induced shift, ΔEF, changes the population of electronic states near the Fermi level. Hence, all transitions between van Hove singularities, whose transition energies (ΔEii) fulfill the following condition:
| ΔEii≤ 2ΔEf |
The actual ΔEF can be quantified by Raman spectroscopy,59,78X-ray photoelectron spectroscopy79 and thermoelectric power measurements.78,80
Pristine CNT are naturally p-doped. SWCNT based FET built from as grown tubes are found to be unipolar p-type, that is, no electron current flows even at large positive gate biases.81
If one succeeds in shifting the Fermi level toward the transport states, this could reduce ohmic losses, ease carrier injection from contacts, and increase the built in potential of Schottky or p–n junctions. Key points to p- and n-type CNT are stability, tuning of doping level and mass production.
In chemical redox doping of SWCNT, the number of oxidants or reductants is restricted to the reagent, which act as pure electron or holes donors without causing irreversible chemical modifications of the SWCNT surface. The charge transfer between the SWCNT and the redox active species is regulated by their ionization energy or electron affinity. In other words, reacting CNT with large electron affinity acceptors affords p-type doping, while a reaction with small ionization energy donors afford n-type doping. Major challenges involve the control of extra charges transferred to CNT and the homogeneity in the bulk sample.
The chemical doping of carbon nanotubes are realized by intercalating electron donors or acceptors, substitutional doping, encapsulating atoms, molecules or clusters gas adsorption, non-covalent functionalization with organic molecules or wrapping of polymers or covalent functionalization.
Intercalating electron donors or acceptors
Doping reactions, namely, intercalation of potassium, rubidium, iodine and bromine in the interstitial channels of SWCNT were performed in the vapor phase using the two-bulb method and studied by Raman spectroscopy.48 The results are consistent with electron transfer from the CNT to the various dopants. A stoichiometry controlled doping with lithium and rubidium shows up as a monotonic upshift of the G-modes, loss of absorption bands and increase of conductivity.51Bromine leads, for example, to a conductivity decrease of up to 30 times, while in the case of potassium the differences are as large as 20.48The electron transfer doping with lithium, iodine, bromine and with the radical anions of naphthalene, fluorenone and anthraquinone were confirmed by absorption spectroscopy. Overall, the conductivity increase and the Fermi level shift are linearly proportional to the difference in redox potential between the SWCNT and the dopant.47 In another experiment, one terminus of individual SWCNT grown on SiO2 was n-doped with potassium. Electron and hole concentrations at these two sides were further tuned by a gate potential and an intratube p–n junction with rectifying characteristics was obtained.82
The intercalation of other metals such as silver into SWCNT bundles has also been studied83 here, the downshift in the G-band indicates that electrons are, however, transferred from the silver atoms to the SWCNT.84
Recently, air-stable, iodine doped SWCNT/polymer composites were prepared. This composite showed enhancement in electrical conductivity by a factor of 2 to 5 when compared to undoped SWCNT/polymer composites.85 Furthermore, Lewis acids (FeCl3,86 WF6,87 CrO3,83SOCl288) and also Brønsted acids (sulfuric, nitric, hydrochloric,78,79,89trifluoroacetic acid,90N-hydroxyheptafluorobutyramide (HFBA), perfluorododecanoic acid (PFDA), 2-methyl acrylamidopropane sulfonic acid (AMPS), aminobutyl phosphonic acid (ABPA), and hexadecyl phosphonic acid (HDPA))91 were investigated for CNT p-doping.
Substitutional doping
The substitutional doping involves the replacement of carbon atoms by boron or nitrogen atoms that, in turn, introduce strongly localized electronic features in the valence or conduction bands, respectively, and enhance the number of electronic states at the Fermi level depending on the location and concentration of dopants.Boron has one electron less than carbon, and when incorporated into SWCNT generates sharp localized states below the Fermi level (valence band). These states are caused by the presence of holes in the structure, and the tube could be considered as a p-type semiconductor. Boron doped SWCNT (B-SWCNT) with boron contents up to 3% were synthesized using laser ablation of graphite-B-Co-Ni targets.92 Other synthesis methods for highly doped B-SWCNT, in which 15% of the carbon atoms are replaced by boron, have been reported.93 Microwave conductivity studies on bulk B-doped MWCNT reveal that these structures are intrinsically metallic.94
One possible structure of N-SWCNT is a three-coordinated nitrogen atom within the sp2-hybridized network, which induces sharp localized states above the Fermi level due to the presence of additional electrons95 (Scheme 5(a)). These N-SWCNT exhibit n-type conduction. The second type of substitutional nitrogen leave two-coordinated nitrogens in the SWCNT lattice—provided that an additional carbon atom is removed from the framework (Scheme 5(b)).
 | ||
| Scheme 5 N-SWCNT with (a) substitutional N and (b) pyridine-type N. | ||
This type of defect induces the presence of localized states below and above the Fermi level. Therefore, either a p- or n-type conductor—depending on the level of doping, the number of nitrogen atoms and the number of removed carbon atoms within the hexagonal sheet—will result.
N-MWCNT with low concentrations of nitrogen (< 1%) were synthesized by arc-discharge in the co-presence of melamine, nickel and yttrium96 or viapyrolysis of pyridine and methylpyrimidine.97 The first observation of aligned arrays of N-MWCNT (<1–2%) involved the pyrolysis of aminodichlorotriazine and triaminotriazine over laser etched cobalt thin films at 1050 °C.98 Long strands of N-SWCNT bundles that were successfully produced by pyrolyzing ferrocene in an argon atmosphere in ethanol solutions with benzylamine at 950 °C give rise to an electron conduction quite different when compared to that of pure carbon SWCNT, especially at temperatures lower than 20 K.99
Bundles of B-SWCNT and N-SWCNT can also be produced using partial substitution in the presence of B2O3 vapor and N2 at 1523–1623 K100 Several groups have also succeeded in synthesizing tubes with a BxCyNz composition, by arc discharge101 and by gas-phase pyrolysis.102
It is also important to mention that besides nitrogen and boron, other elements such as silicon103 and phosphorous104 could also dope CNT.
Doping by encapsulating atoms, molecules or clusters in the tube interior space (endohedral doping)
The filling of carbon nanotubes with single elements, empty fullerenes, endo-, and exohedral metallofullerenes, exohedral fullerene derivatives, alkali metal, metal halides and hydroxides, transition metal oxide and complexes, etc. has been a very active field.14j,105 The endohedral guest are viewed as dopants that, in turn, help to gain control over the nanotube properties such as conductance106 and/or electronic band gap.107 It has been demonstrated that, for example, periodic arrays of C60 in SWCNT give rise to a new hybrid electronic band.108 The resulting increase in CNT conductivity is based on electron transfer that occurs from SWCNT to C60 leaving holes as the primary charge carriers behind.109 Important insights into the electronic structure of the encapsulated C60, C82, La@C82, and Gd@C82110 were lent from Raman spectroscopy. In particular, dramatic changes relative to empty SWCNT are seen:111 a sharp and intense line, for example, at 142 cm−1 has been interpreted as a polymerization signature of the encapsulated metallofullerenes. Additional signals appeared at 400, 520 and 640 cm−1. The upshift (stiffening) of the G-band suggest also in this case a charge transfer process from SWCNT to the encapsulated fullerene.112Interestingly, encapsulation of metallocenes like bis(cyclopentadienyl)cobalt and bis(ethylcyclopentadienyl) cobalt inside SWCNT, has led to hybrid systems with modified electronic and optical properties. Electron transfer from the cobalt ions to the SWCNT is implied when considering the change in the charge state of the encapsulated molecules. The filling of the tubes induces a red-shift of the photoluminescence emission, which is attributed to the formation of localized states that are located below the conduction band of SWCNT.113
A variety of organic molecules with different ionization energies and electron affinities (i.e., anthracene, tetracene, pentacene, tetracyanoquinodimethane (TCNQ), tetrafluorotetracyano-p-quinodimethane (F4TCNQ), tetrakis(dimethylamino)ethylene (TDAE), tetrathiafulvalene (TTF), tetramethyl tetraselenafulvalene (TMTSF), 3,5-dinitrobenzonitrile) encapsulate within the interior of SWCNT.114 Depending on the electron affinity and/or ionization potential of the dopant, the resulting SWCNT turn out to be either p-type or n-type semiconductor. The resistivity of TCNQ@SWCNT, TDAE@SWCNT, TTF@SWCNT, TMTSF@SWCNT and F4TCNQ@SWCNT was approximately 50% of that found for pristine SWCNT and was with the exception of TDAE@SWCNT stable under air. A pronounced intratubular charge transfer evolves around TCNQ@SWCNT, which contrasts, nevertheless, the lack of charge transfer activity in C60@SWCNT. Note that TCNQ and C60 have approximately the same electron affinity.114,115
Doping by gas adsorption
The CNT electrical resistance, thermoelectric power, and local density of states, as determined by transport measurements and scanning tunneling spectroscopy are extremely sensitive to exposure to air or oxygen. Such parameters are reversibly tuned by surprisingly small concentrations of adsorbed gases. An impressing demonstration of such alterations is that an intrinsically semiconducting nanotube converts into an apparent metallic one once exposed to air/oxygen.68c,80a,116Annealing the contacts of a SWCNT FET in vacuum117 or in an inert gas,118 removes the adsorbed oxygen and reconverts the device from p- to n-type. n-type doping of SWCNT by ammonia adsorption was also observed.119 Smaller are the effects when SWCNT are in contact with H2, He and N2. Simply, weaker interactions are responsible for such trends.120Doping by noncovalent functionalization with organic molecules or wrapping of polymers
Molecular physisorption as it affects the electrical properties of SWCNT is of fundamental interest and importance to potential molecular electronics and sensors.22a,68c,121SWCNT that were suspended in aqueous SDS suspension were tested with different electron acceptors—7,7,8,8-tetracyanoquinodimethane (TCNQ), 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (TFTCNQ), mordant yellow 10 (MY) and 4-amino-1,1-azobenzene-3,4-disulfonic acid (AB)—as charge transfer agents. Time resolved emission spectra attest that the intensity loss starts with the tubes of lowest energy band gap and largest diameter and progresses with time to the largest band gap/smallest diameter semiconductors. Common is this trend for all the mentioned electron acceptors. They differ, only, in rate and effectiveness of charge transfer.122 While TCNQ increased the CNT conductivity, adsorption of TTF onto the nanotube surface led correspondingly to a decrease.123
Notably, adsorption of polymers such as polyethyleneimine evokes n-doping of SWCNT and to stabilize the doped state. A 10 cm−1 downshift of the G-band confirm the electron transfer from the polymer to the CNT surface.124Nafion, on the other hand, exerted to SWCNT in a solvent mixture containing deionized water and 1-propanol a p-type behavior. Subsequent treatment with nitric acid removed the Nafion layer and de-doped the SWCNT. Nevertheless, acid-treated SWCNT/Nafion is not so sensitive to environmental changes.125
Electrical transport measurements reveal that the electrical conductance of SWCNT changes drastically upon adsorption of butylamine and 3′-(aminopropyl)triethoxysilane. The amine groups are electron donors and responsible for the charge transfer, and, thus, the observed electrical conductance change. Adsorption of amines causes modulated chemical gating and intramolecular wire junctions that exhibits pronounced rectifying diode behavior. The physisorbed amines desorb typically within 12 h and the nonrectifying characteristics are restored. Cross-linking of heterofunctional 3′-(aminopropyl)triethoxysilane on partially exposed nanotubesviasiloxane bridges however fixes intrananotube rectifying behavior.126
Electron acceptor/electron donor in donor–acceptor systems
CNT that are modified by photoactive molecules are very actively studied as donor–acceptor nanohybrid models and as building blocks in optoelectronic devices.127Electron acceptor in donor–acceptor systems
The carboxylated CNT defect sites are often transformed into amine or ester by amination or esterification. The most widely studied photoactive components bound to CNT are porphyrins known to be good light harvesting chromophores and/or excited state electron donors.128Scheme 6 documents how photo- and redox-active porphyrins have been linked to the carboxylate functionalities through esterification of SWCNT bound carboxylic acid. FTIR and thermal analysis indicated that amide or ester bonds were formed to bridge between the excited state electron donor and SWCNT. In the corresponding electron donor–acceptor nanoconjugates (i.e., SWCNT–H2P), the photoexcited porphyrins deactivate through a transduction of excited state energy. Interestingly, the rates and efficiencies of the excited state transfer depend on the length of the tether that links the porphyrins with the SWCNT. To this end materials with shorter tethers showed the least fluorescence quenching.129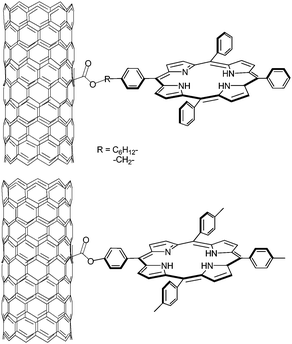 | ||
| Scheme 6 Synthesis of SWCNT bearing porphyrins. | ||
Similarly, phthalocyanines have drawn considerable attention as molecular materials with outstanding electronic and optical properties (i.e., broad light absorption combined with high photo- and chemical stability).128d,130 Unsymmetrically-substituted aminophthalocyanines (ZnPc) react with SWCNT at the terminal carboxylic acid groups of shortened, chemically-etched samples.131 However, the resulting materials are nearly insoluble in common organic solvents. The solubility/dispersability of the SWCNT–ZnPc conjugates is, however, an indispensable task for a ready manipulation and a feasible solution phase processing.
Applying the same protocol was successful to integrate very strong electron donors like tetrathiafulvalene (TTF) and extended tetrathiafulvalene (exTTF)—see Scheme 7.132 A detailed photophysical investigation supports the occurrence of photoinduced electron transfer processes. Importantly, inserting spacers of different length fine controls the rate of the electron transfer.133
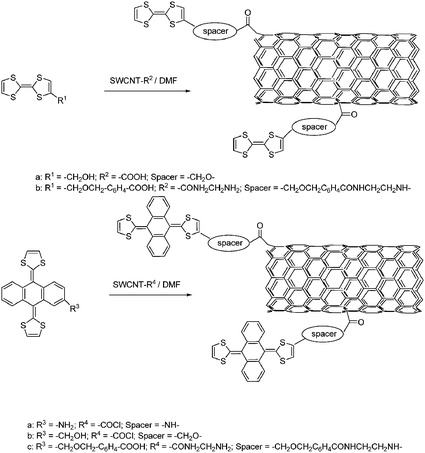 | ||
| Scheme 7 Synthesis of SWCNT bearing TTF and exTTF. | ||
CNT amides or esters are important precursors in the context of various fluorescent dyes—for a summary of leading examples see Scheme 8.1h,134
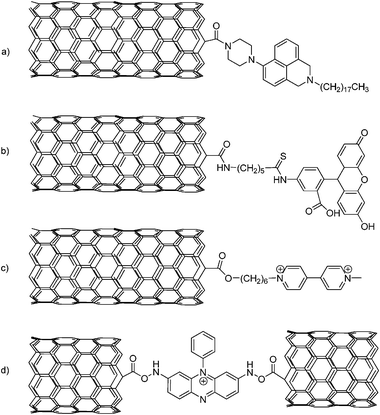 | ||
| Scheme 8 Some leading examples of fluorescent dyes that are covalently attached to SWCNT: (a) naphthalimide, (b) 5-(5-aminopentyl)thioureidyl fluorescein, (c) asymmetrically substituted viologen and (d) phenosafranin (a cationic dye PS+, 3,7-diamino-5-phenylphenazinium). | ||
Importantly, ruthenium(II) bipyridine complexes that act as photoexcited state electron donors to CNT when linked to them135 are integrative components for dye sensitized solar cells (DSSC)136 or as a tool for controlling the conductivity of CNT in molecular electronics.137 On the other hand, the reversible coordination of copper(II) terpyridine afforded a thermally stable, neutral nanocomposite possessing notable luminescence properties (see Scheme 9).138
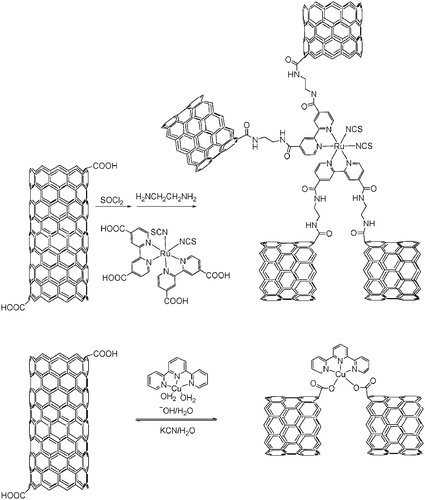 | ||
| Scheme 9 Synthesis of SWCNT bearing polypyridyl complexes. | ||
Carboxylate chemistry permitted also the immobilization of tailorable metallic and/or semiconducting nanoparticles onto the surface of CNT. One very successful strategy aims at tethering gold nanoparticles, for example, to SWCNT and MWCNT. This strategy encompasses the treatment of oxidized SWCNT, namely, thiol-derivatized, by reacting them with NH2–(CH2)n–SH.139 A different strategy involves the reduction of carboxylates at the open ends of CNT, their chlorination and subsequent thiolation. The corresponding thiol groups were afterward conveniently assembled to silver140 and gold nanoparticles,141 or even to cadmium sulfide (CdS) nanoparticles.142 Oxidized SWCNT and MWCNT react with carboxylic acid terminated cadmium selenide (CdSe) nanocrystals that are capped with mercaptothiol derivatives.143 Similarly, titanium dioxide (TiO2) nanocrystals, functionalized with 11-aminoundecanoic acid, were probed. Hereby, the aid of intermediary linking agents, which were either ethylenediamine or semicarbazide, was extremely beneficial to guarantee the integrity of the nanoscale heterostructured materials.144 Similarly, oxidized MWCNT interact with thiol-stabilized ZnS-capped CdSe quantum dots containing amine terminal groups.145
Moreover, SWCNT that are oxidized by either acid or ozone treatment assemble quite efficiently onto amine modified gold surfaces.139e,146 The corresponding electrodes are ideally suited to investigate the charge transfer process between SWCNT and the underlying substrates.146a When the oxidized ends of CNT are functionalized asymmetrically147 onto gold and mercury surfaces the modulation of current rectification has been recently demonstrated.148
By cycloaddition of azomethine ylides—generated in situ by the condensation of α-amino-acids and aldehydes—several electron donors were attached to the sidewalls of CNT. In this context, ferrocene149—see Scheme 10(a)—and tetraphenylporphyrins150 bridged by a second generation of polyamidoamine (PAMAM) dendrimers—see Scheme 10(b)—are leading examples. In the latter case, the dendrimers are linked directly to the SWCNT surface using a divergent methodology. This approach allows increasing the number of functional groups on the nanotubes without provoking significant damages to the conjugated π-system of SWCNT. The photophysical properties of the resulting nanoconjugates (i.e., SWCNT–(H2P)n) have been investigated with a series of steady state and time resolved spectroscopic methods. The fluorescence kinetics, for example, provides evidence for two transient decays: one very short-lived (i.e., 0.04 ± 0.01 ns) and one long-lived (i.e., 8.6 ± 1.2 ns). A plausible explanation is that some porphyrin units do not interact with the nanotubes, thus exhibiting a fluorescence lifetime similar to that of the free porphyrin. Complementary transient absorption measurements not only corroborate the fast decay of the photoexcited H2P, but also confirm that intraconjugate charge separation evolves from the excited porphyrin to the SWCNT.
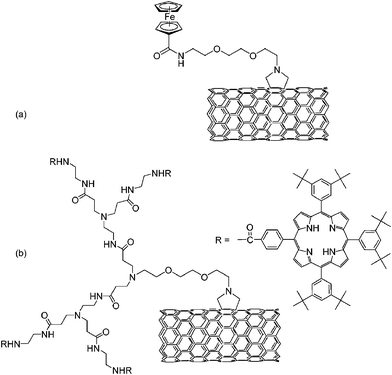 | ||
| Scheme 10 Amidoferrocenyl (a) and PAMAM bridged tetraphenyl porphyrin (b) SWCNT nanoconjugates. | ||
Additional examples are (see Scheme 11) free base phthalocyanines (H2Pc)151 and zinc phthalocyanines (ZnPc)152 that were grafted via1,3-dipolar cycloaddition of azomethine ylide to SWCNT.
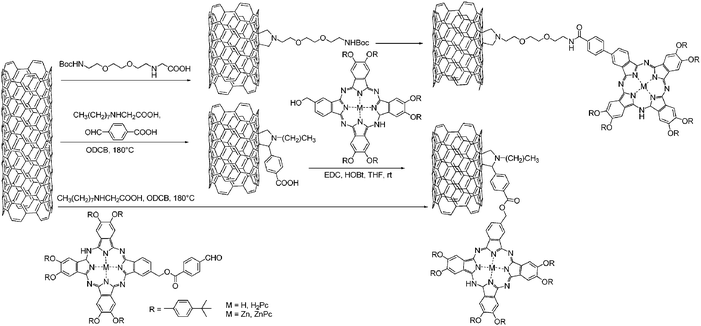 | ||
| Scheme 11 H2Pc–SWCNT and ZnPc–SWCNT nanoconjugate by 1,3 dipolar cycloaddition. | ||
Similar but not identical is the use of 1,3-cycloaddition of nitrile oxide to ester modified SWCNT. In particular, pyridyl isoxazolino units were added along the CNT sidewalls—Scheme 12.153 The attached pyridines form complexes with ZnP as established by electrochemical studies. Upon photochemical excitation, mainly energy transfer funnels the light from the singlet excited porphyrins to SWCNT.
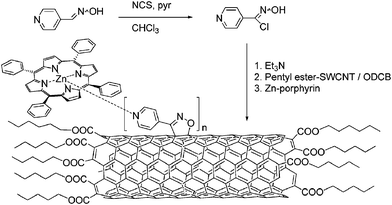 | ||
| Scheme 12 1,3-Cycloaddition of nitrile oxide and formation of SWCNT–ZnP nanoconjugates. | ||
Functionalization with aryl diazonium salts, generated in situ is another effective method for CNT sidewall grafting.14j What is particularly interesting is that the metallic SWCNT seem to react more rapidly than the semiconducting analogous. The selectivity is dictated by the availability of electrons near the Fermi level to stabilize a charge transfer transition state preceding the bond formation. This opens the way for a selective solubilization and separation. In the last step, UV laser irradiation might remove these aryl moieties and, in turn, facilitates the recovery of purified—but yet pristine—SWCNT.154In situ generated tetraphenylporphyrin diazonium salts react directly with SWCNT. These functionalized SWCNT show superior optical limiting effects when compared with pristine SWCNT—see Scheme 13.155
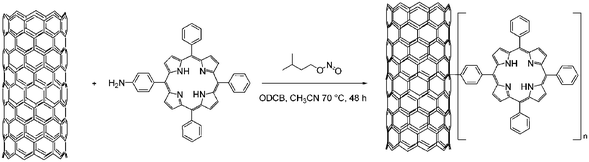 | ||
| Scheme 13 SWCNT–H2P nanoconjugates. | ||
Still an important consideration when associating CNT with electron donor building blocks is to preserve as much as possible the unique electronic structures of the CNT. A versatile approach involves grafting SWCNT with polymers such as poly(sodium 4-styrenesulfonate) (SWCNT–PSSn−) with a SWCNT to PSSn− ratio of 55:45—see Scheme 14(b).156 In turn, highly stable water dispersable SWCNT were prepared with relative ease. The attached PSSn− functionalities also assist in exfoliating individual SWCNT–PSSn− from the larger bundles. AFM and TEM analysis corroborated the presence of SWCNT with lengths reaching several micrometers and diameters around 1.2 nm.
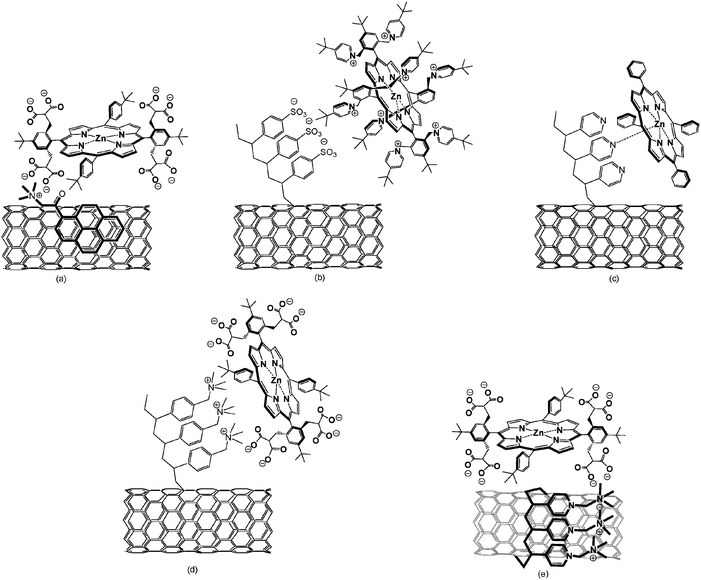 | ||
| Scheme 14 SWCNT based nanohybrids: (a) SWCNT/pyrene+/ZnP8−; (b) SWCNT–PSS−/ZnP8+; (c) SWCNT–PVP/ZnP; (d) SWCNT–PVBTA/ZnP8−; (e) SWCNT/PVBTA/ZnP8−. | ||
A Coulomb complex formation was achieved between SWCNT–PSSn− and an octapyridinium H2P salt (H2P8+)—see Scheme 14(b). Several spectroscopic techniques such as absorption, fluorescence and TEM were used to monitor the complex formation between SWCNT–PSSn− and H2P8+, yielding SWCNT–PSSn−/H2P8+. Importantly, photoexcitation of H2P8+ results in the newly formed nanohybrid in an efficient intrahybrid charge separation event (0.3 ns). The charge separation is governed by a large thermodynamic driving force of 0.81 eV. The newly formed radical ion pair exhibits a remarkably long lifetime of 14 μs under anaerobic conditions, which constitutes one of longest reported for any CNT ensemble found so far.
Similarly, dispersable SWCNT—grafted with poly(4-vinylpyridine) (PVP)—were assayed in coordination tests with ZnP—see Scheme 14(c).157 Grafting of the polymer to the SWCNT sidewalls was supported by Raman and near-IR spectra and the composition of the resulting SWCNT–PVP was estimated as 61/39. Kinetic and spectroscopic evidence corroborates the successful formation of SWCNT–PVP/ZnP nanohybrids in solutions. Within this SWCNT–PVP/ZnP nanohybrid, static electron transfer quenching 2.0 (±0.1)×109 s−1 converts the photoexcited ZnP chromophore into a microsecond-lived radical ion pair state; that is, one-electron oxidized ZnP and reduced SWCNT.
Reacting poly((vinylbenzyl)trimethylammonium chloride) (PVBTAn+) through free-radical polymerization of the monomer in the presence of SWCNT yielded polymer grafted SWCNT. A donor–acceptor nanohybrid have been afterward prepared using electrostatic/van der Waals interactions between covalent SWCNT-PVBTAn+ and 5,15-bis[2′,6′-bis{2″,2″-bis(carboxy)ethyl}methyl-4′-tert-butylphenyl]-10,20-bis(4′-tert-butylphenyl)porphyrin octasodium (H2P8− and/or ZnP8−)—see Scheme 14(d).158
SWCNT/PVBTAn+ are water suspendable SWCNT, in which PVBTAn+ are non-covalently wrapped around them. For an illustration see Scheme 14(e). Versatile donor acceptor nanohybrids have been prepared using electrostatic/van der Waals interactions between SWCNT/PVBTAn+ nanohybrids and porphyrins (H2P8− and/or ZnP8−). Several spectroscopic, microscopic, transient and photoelectrochemical measurements were employed to characterize the resulting supramolecular complexes. The photoexcitation of the nanohybrids afforded long-lived radical ion pairs with lifetimes as long as 2.2 μs.158
Polycyclic aromatic hydrocarbons (i.e., pyrene159 or naphthalene160) that bear ionic, namely, anionic and cationic functionalities are similarly suited to suspend SWCNT. Crucially, the interactions between CNT and the adsorbates have also been attributed to charge transfer. Water-soluble SWCNT were obtained in aqueous solutions of 1-(trimethylammoniumacetyl)pyrene (pyrene+) (see Scheme 14(a)).161 Decisive evidence for SWCNT/pyrene+ interactions came from fluorescence experiments, which showed the quenching of pyrene+ fluorescence and amplification of fine structure in the near-visible region due to the successive removal of free pyrene+.162 The solubility of CNT in the resulting black suspensions is as high as 0.2 mg ml−1 and the stability reaches several months under ambient conditions without showing any apparent precipitations. TEM and AFM images revealed the coexistence of very thin bundles of SWCNT/pyrene+, 1–3 μm in length and 3–20 nm in diameter, and individual SWCNT/pyrene+ with diameters as small as 1.3 nm.163 Decisive information about excited state interactions came from transient absorption measurements, where the spectral changes in the 400–600 nm region reflect the features associated with pyrene+ excited state, while the changes in the 600–800 nm region correspond to SWCNT. Now that the surface of SWCNT is covered with positively charged ionic head groups, van der Waals and electrostatic interactions are utilized to complex oppositely charged electron donors. Water-soluble porphyrin salts (H2P8−) and the related zinc complexes (ZnP8−) were selected as ideal candidates for the development of SWCNT-based photoactive systems—see Scheme 14(a).164 The supramolecular association was followed by spectroscopic (absorption and fluorescence) and microscopic (TEM and AFM) means. In absorption experiments, the successful complex formation—for instance, of CNT and ZnP8−—were confirmed by red-shifted Soret- and Q-bands and the development of a series of isosbestic points. In the composite systems, fluorescence and transition absorption studies showed rapid intrahybrid electron transfer (0.2 ± 0.05 ns). The differential absorption changes of the SWCNT/pyrene+/ZnP8− or SWCNT/pyrene+/H2P8−assemblies are governed by broad absorptions in the visible between 600–800 nm because of ZnP8− or H2P8− centered redox products.
As alternatives water soluble electron donors like a copolymer of unsubstituted thiophene and 7-(thien-3-ylsulfanyl)heptanoic acid (PSCOOH)165 and size-quantized thioglycolic acid stabilized cadmium telluride (CdTe@TGA) nanoparticles166 emerged, which were combined with SWCNT/pyrene+—see Scheme 15. In the resulting nanohybrids strong electronic interactions were noted. In particular, polythiophene and/or CdTe tend to donate an excited state electron to the ground state of SWCNT.
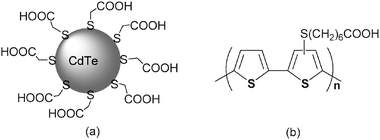 | ||
| Scheme 15 CdTe@TGA and PSCOOH that interact with CNT via non-covalent means. | ||
Stable SWCNT/H2P composites were also realized when condensing tetraformylporphyrins and diaminopyrenes onto SWCNT.167 The degree of interaction between SWCNT and H2P polymer was evaluated by absorption and fluorescence spectra, and chemical removal of H2P from SWCNT. In SWCNT/H2P, the Soret and Q bands of H2P moieties were significantly broadened and their fluorescence was almost completely quenched.
A highly soluble, conjugated ZnP-polymer was synthesized and found to immobilize onto the surface of SWCNT, producing a soluble SWCNT/ZnP-polymer complex. Successful complexation required, however, the addition of trifluoroacetic acid.168 The SWCNT/ZnP-polymer assembly resulted in enhanced planarization and conjugation within the porphyrin polymer, which was manifested in a 127 nm bathochromic shift of the Q-band absorption. Control experiments with the ZnP-monomer indicated that homogeneous solutions could be prepared by means of sonication, but the SWCNT/ZnP-monomer interactions were significantly weaker, leading to SWCNT precipitation within minutes. Similarly, a novel triply fused ZnP-trimer—Scheme 16—gives rise to extremely strong and nearly irreversible supramolecular interactions with SWCNT, resulting in stable solutions of SWCNT/ZnP-trimer in THF.169
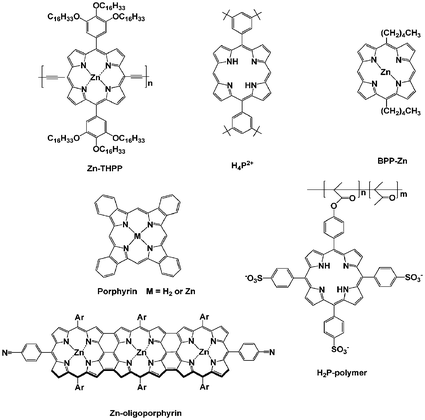 | ||
| Scheme 16 Porphyrin derivatives that interact with CNT via non-covalent means. | ||
An impressive SWCNT solubility has been achieved upon treatment with tetraphenylporphyrin (i.e., H2P and ZnP) in DMF.170TEM and AFM images revealed the existence of high density SWCNT and exfoliation of individual SWCNT as a result of the tedious work up procedure. The CNT diameter ranged typically from 0.9 to 1.5 nm, underlining the successful debundling properties of ZnP. Evidence for interactions between porphyrins and SWCNT was obtained from fluorescence spectra, where the porphyrin fluorescence was significantly quenched compared to that of the bare porphyrin. The fluorescence quenching has been ascribed to energy transfer between the photoexcited porphyrin and SWCNT.
In a similar fashion SWCNT were non-covalently functionalized with porphyrins (i.e., H2P and ZnP).171 The absorption and fluorescence spectra of SWCNT/H2P showed efficient electronic communications between the π-systems of SWCNT and H2P, while weaker interactions were observed for SWCNT/ZnP. Clear evidence for π–π interactions between SWCNT and H2P were derived from AFM measurements. The AFM image of SWCNT/H2P revealed smaller bundles than SWCNT/ZnP with some individual CNT (diameter 1.5 ± 0.2 nm). Moreover, the SWCNT/H2P suspensions are more stable than those of SWCNT/ZnP.
Water-soluble H2P [meso(tetrakis-4-sulfonatophenyl)porphine dihydrochloride] polymers were exploited to suspend SWCNT in aqueous solutions.172 Owing to interacting H2P and SWCNT the suspensions are stable for several weeks and protect the free base against protonation to the corresponding diacid. Under mildly acidic conditions SWCNT mediated J-aggregates are formed, which are, however, unstable in solution. Consequently, the precipitation of SWCNT occurs over the course of a few days.
Suspending SWCNT in the presence of H2P—[5,10,15,20-tetrakis(hexadecyloxyphenyl)-21H,23H-porphine] (Zn-THPP)—in organic solvents yielded novel soluble SWCNT/H2P.173 The insoluble and recovered SWCNT were separated from H2P by treatment with acetic acid and vigorous centrifugation. After heating the recovered SWCNT and the free SWCNT to 800 °C in a nitrogen atmosphere, the spectroscopic analysis showed that the semiconducting SWCNT and the free SWCNT are enriched in recovered and metallic SWCNT, respectively. Under ambient conditions, the bulk conductivity of the semiconducting SWCNT (i.e., recovered) is 0.007 S cm−1, while that of the metallic SWCNT (i.e., free) is 1.1 S cm−1. Thus, selective interactions between H2P and semiconducting SWCNT are the inception to a successful separation of metallic SWCNT and semiconducting SWCNT.
Electron donors in donor–acceptor systems
To this date, fewer examples of nanohybrids, where CNT function as a photoinduced electron donor, are known. One of the few is the electron transfer system that is based on SWCNT and [5,15-bis(4-pyridyl)-2,8,12,18-tetraethyl-3,7,13,17-tetramethyl zinc porphyrin]—see Scheme 17(a).174 Field effect transistor characteristics suggests that the rate and magnitude of the process depends both on the wavelength and on the intensity of applied light, with a maximum value of 0.37 electrons per porphyrin upon 420 nm illumination with 100 W m−2.![(a) SWCNT/[5,15-bis(4-pyridyl)-2,8,12,18-tetraethyl-3,7,13,17-tetramethyl zinc porphyrin]; (b) SWCNT/Pyrene-NH3+/crown-C60; (c) SWCNT/sapphyrin.](/image/article/2009/CS/b802652c/b802652c-s17.gif) | ||
| Scheme 17 (a) SWCNT/[5,15-bis(4-pyridyl)-2,8,12,18-tetraethyl-3,7,13,17-tetramethyl zinc porphyrin]; (b) SWCNT/Pyrene-NH3+/crown-C60; (c) SWCNT/sapphyrin. | ||
A more recent demonstration aimed at designing an alkyl ammonium functionalized pyrene (pyrene-NH3+). This not only interacts with SWCNT, but also complexes benzo-18-crown-6. Such ammonium/crown ether interactions were used to associate C60 (i.e., crown-C60) to yield stable SWCNT/pyrene-NH3+/crown-C60 nanohybrids (see Scheme 17(b)). Steady-state and time-resolved absorption spectroscopy prompted to a photoinduced electron transfer, by which SWCNT and C60 are oxidized and reduced, respectively. The lifetime of the newly formed charge separate state was on the order of 100 ns.175
Scheme 17(c) displays the final example, namely, a supramolecular complex between SWCNT and a functionalized sapphyrin diol macrocycle. An impressive benefit is the SWCNT solubilization in water and in 1-butyl-3-methylimidazolium hexafluorophosphate (BMIM-PF6). In these solvents, proofs of a photoinduced electron transfer from SWCNT to the sapphyrin moiety came from a combination of steady-state investigations, femtosecond transient absorption spectroscopies and pulse radiolysis experiments.176
Integrative components in photoelectrodes/photoanodes and photocathodes
In recent years the continuing interest in alternative photovoltaic technologies, especially with respect to lowering production costs, triggered extensive research in the fields of organic photovoltaic (OPV) devices. Compared with inorganics materials, organic materials offer great incentives including device production at exceptionally low costs, lightweight and flexible devices that enabling versatile applications.177In this context, organic materials absorb light in most of the visible and near-infrared regions of the solar spectrum to generate conduction band electrons and valence band holes. Coulombic interactions between the spatially confined charges result in strongly bound excitons. These excitons must, however, be separated, at the interphases between the electron donors and the electron acceptors or between the photoactive materials and semiconducting electrodes, before they recombine or relax in alternative fashions to the ground state.178 Finally, the resulting free charge carriers (i.e., electrons and holes) should be transported to an external circuit, while minimizing recombination pathways. To this end, the main obstacles to realize widespread applicability of organic semiconductor for OPV are: high exciton binding energy, low charge carrier mobility and susceptibility to degradation. All these factors impact adversely the device efficiency and its lifetime.
Relative to known organic materials, CNT bear the great advantage to be structurally robust, chemically stable and highly conductive. They can improve exciton dissociation in the presence of an external field at the heterointerfaces. Their high surface area—of ca. 1500 m2 g−1—favors large donor–acceptor contact surfaces within the photoactive layer.179 All of these aspects are very important to separate the aforementioned excitons. Also the tubular shape of CNT appears interesting. This in particular, should enable a lower percolation threshold for the acceptor phase and consequently a more efficient charge transport to the external circuit.180
Thin film photovoltaics
Pioneering studies have revealed rectifying heterojunctions that are formed between poly(2,5-bis(cholestanoxy)-1,4-phenylene vinylene) and MWCNT and that are photosensitive.181 Since these initial studies much research efforts has focused on SWCNT-182 or MWCNT-conjugated polymer composites,183 sometimes with the co-presence of C60184, CdSe185 or phthalocyanine.182cTypically, CNT are dispersed in solutions containing the electron donating polymer—typically poly(3-hexylthiophene) (P3HT) or poly(3-octylthiophene) (P3OT). The obtained blends are spin coated onto transparent conductive electrodes in order to obtain a bulk heterojunction film having thicknesses that range from 60 to 120 nm. Such electrodes are glass slides covered with indium tin oxide (ITO). A thin 30–70 nm sublayer of (poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) between ITO and photoactive layer favors selective hole injection into ITO electrode, helps in smoothing the ITO surface, reduces the density of pinholes and suppresses current leakage effects that arise around shunting paths. Finally, a cathode consisting of a 20–70 nm thick layer of aluminium—sometimes with an intermediate layer of lithium fluoride—is thermally evaporated and/or sputtered on top of the photoactive material. Post fabrication treatment such as heating to the point beyond the glass transition temperature of either P3HT or P3OT is beneficial for manipulating the phase separation of the blend. Since the polymers are microcrystalline systems it also affects the ordering of the polymeric chains. Overall, this annealing improves charge transfer, charge transport, and charge collection throughout the device.186 As a consequence of such ordering, the hole mobility, and hence the power efficiency of the polymer CNT device, increases significantly.187 So far, following this strategy a maximum power conversion efficiency (η) of 0.22% has been obtained.188 Although the polymer CNT cells give rise to an exceptionally high open circuit voltage between 0.75 and 1 V as well as an advantageous near infrared light harvesting,189 their photocurrents are quite low.190 Please compare, for example, photocurrents of 0.25 to 0.5 mA cm2 to 10.6 mA cm2 measured for an optimized ITO/PEDOT:PSS/P3HT:[6,6]-phenyl-C61-butyric acid methyl ester (PCBM)/Al OPV device.191
Quantum mechanical ab initio calculations were run to provide insights into the heterojunction interfaces between CNT and P3HT. In the case of the semiconducting SWCNT, the results show small interactions between CNT and the polymer, which forms heterojunctions with an interface desirable for photovoltaic. However, when considering the metallic SWCNT, appreciable charge transfer was found to prevail from P3HT, in its ground state, to CNT. This enhances the susceptibility of P3HT for negative charges at the interface. Implicit is an electron transfer to the CNT, which is, nevertheless, counter-productive during the step of exciton dissociation.192 These studies explain the moderate photocurrents observed for the P3HT/CNT photoactive blends, compared to, for example, P3HT/C60. This incomplete exciton dissociation dominates at low CNT concentrations—about 1 wt% for SWCNT193 and 5 wt% for MWCNT.194 Suspending, on the other hand, higher percentages of CNT within the polymer matrix, decreases the efficiency. This trend is rationalized on the basis of short circuits, since the CNT length is comparable with the overall thickness of the photoactive layer. Finally, the presence of CNT ropes/bundles and, moreover, appreciable amounts of metallic entities should be considered. All these rationales decrease the carrier selectivity (i.e., holes and electrons) in the device and favor the recombination processes.
In a recent publication an impressive power conversion of 4.9% was obtained in a modified ITO/PEDOT:PSS/P3HT:PCBM/Al solar cell, after depositing a SWCNT layer between the ITO and the PEDOT:PSS or between the PEDOT:PSS and the photoactive blend. Here, SWCNT were deposited by dip-coating from a hydrophilic suspension after an initial argon plasma treatment of the surface.195
Photoelectrochemical cells
Some promising reports on photoelectrochemical devices integrating SWCNT—as electron acceptor—on ITO are based on the layer by layer strategy (LbL). The films are formed by dip coating, that is, depositing alternating layers of oppositely charged materials. In most of these instances the ITO electrodes were covered with a base layer of polyelectrolyte (i.e., poly(diallyldimethylammonium chloride) (PDDAn+) or sodium poly(styrene-4-sulfonate) (PSSn−)) through hydrophobic forces.165,166,196SWCNT/pyrene+ and finally water-soluble but oppositely charged porphyrins (i.e., ZnP8−) were assembled electrostatically—see Scheme 14(a). Lately, this approach has been applied to DWCNT, MWCNT and thin-MWCNT. Among the tested CNT, thin-MWCNT showed the best performance as electron acceptor layer material with monochromatic power conversion efficiencies of up to 10.7% for devices that contain single layers of thin-MWCNT and single layers of ZnP8−.196Similarly, PSCOOH—Scheme 15(b)—were integrated together with SWCNT/pyrene+ onto photoactive ITO electrodes by van der Waals and electrostatic interactions. PSCOOH functions in the device as the light harvesting chromophore that donates electrons to the electron accepting SWCNT. Quite remarkably, the monochromatic incident photoconversion efficiencies for single and eight sandwiched layers of PSCOOH were determined as 1.2 and 9.3%, respectively.165
With the same assembly method, PDDA/SWCNT-PVBTAn+/ZnP8− or PDDA/SWCNT/PVBTAn+/ZnP8−—Scheme 14(d) and (e)—were layered onto ITO substrates. To this end, photoelectrochemical characterization showed IPCE values that maximized at 3.32% and 5.80%, respectively.158
Organic/inorganic composite materials, in which the inorganic semiconductors play a key role for managing charge transfer, were also tested. Very exciting is the use of size quantized CdTe quantum dots (QD).166,197 In particular, water-soluble positively or negatively charged green (2.4 nm), yellow (3.4 nm), and red QD (5.0 nm) have been examined, carrying either L-cysteine or thioglycolic acid—Scheme 15(a)—as surface stabilizers. These surface stabilizers have been beneficial for interactions with pyrene+ immobilized onto CNT. Highest monochromatic IPCE, of up to 2.3%, were registered for hybrid cells consisting of single SWCNT/pyrene+/red-emitting CdTe stacks.166
Another interesting assembly strategy relies on electrophoresis.198 Exposing, for example, SWCNT that were suspended by tetraoctylammonium bromide in tetrahydrofuran to an electrophoretic field assists in depositing them onto ITO covered with SnO2.199 Considerable photoconversion efficiencies were achieved when SWCNT were deposited and assembled with light harvesting CdS QD and/or porphyrins with maximal values of 0.5% and 6.5%, respectively.200 Again, electrophoresis was the key technology to deposit a mixture of 8-aminopentadecane functionalized SWCNT and C60clusters on SnO2. An impressive maximum IPCE of 18% was achieved when a potential of 0.05 V versusSCE was applied.201
A ruthenium dye SWCNT nanoconjugate—Scheme 9—suspended in an ethanolic solution was attached to the TiO2 surface by dip casting. Current voltage characteristics, evaluated in a photoelectrochemical cell, revealed a 0.1 V increase in the open circuit voltage with essentially no change in the short circuit photocurrent, when compared to a reference cell that was soley built on the ruthenium dye. Still, the cell performance (η = 0.34%) is much lower than that of typical Ru dye-based DSSC (η = 10%). Such an increase in open circuit voltage was attributed to the NH groups present in ethylenediamine. A change in basicity of the TiO2 surface results in the negative shift of the conduction band of TiO2. The role of SWCNT as a light scattering layer was also suggested.135b
A different study demonstrated the use of SWCNT networks deposited by electrophoresis on a conducting electrode surface as scaffolds for dye sensitized TiO2 particles to promote charge transport. The TiO2 paste was spread on the ITO/SWCNT/TOAB electrode by the doctor blade technique. After annealing in air, the electrodes were immersed into an ethanol solution of Ru(2,2′-bipyridine)2(2,2′-bipyridine-4,4′-dicarboxylic acid)2+ (Ru(bpy)2(dcbpy)2+). Such an electrode revealed an IPCE enhancement of 1.4 when compared to that lacking SWCNT, due to the suppression of the charge recombination and the improvement of the electron transport.202 Utilizing CdS QD instead of Ru(bpy)2(dcbpy)2+ and the co-presence of a sub layer of SWCNT turned out to be beneficial leading to power conversion efficiencies (1.86%) that increase relative to the electrode lacking SWCNT (1.24%).203
Electrode materials
There is a huge market for transparent electronic conductors such as indium tin oxide (ITO). Between plenty of applications also solar cells necessitate the implementation of conducting, flexible and transparent electrodes. The target for OPV devices is a surface resistivity ≤ 50 ohm sq−1 and a transmittance ≥85% in the solar spectrum.204While extensively developed and optimized, ITO bears, nevertheless, a number of deficiencies. One disadvantage results from its high deposition temperature of around 600 °C, which renders a compatibility with, for example, flexible substrates more than just problematic. Conductivities of ITO on polyethylene terephthalate are about five times lower than that on glass. In addition, cracks appear after repeated bending or strain, and it is not resistant to acid.205 What is even more puzzling is that traditional ITO has poor mechanical properties. Of economical concern is that high quality ITO is expensive.
SWCNT films on polyethyleneterephthalate do not crack after bending, while ITO films become insulating.206 Their high thermal conductivities207 to tolerate heat dissipation and their high radiation resistance—even when irradiated in air—are also particularly striking. Its high optical transparency in a very broad spectral range that starts in the ultraviolet and visible and that extends deep into the near infrared range (3–5 μm) is another benefit. Notable, many transparent conducting coatings are transparent in the visible part of the spectrum, but only a few materials retain good transparency in the infrared while maintaining good electrical conductivity.208
Concluding remarks
To achieve meaningful improvements in the performance of CNT based solar cell, it is highly desirable to use sorted semiconducting SWCNT. Particularly crucial is in this context the energetic location of the conduction band. A controlled cutting of these SWCNT appears also to be necessary, especially to reduce shunting effects in the devices. Finally, deposition methods and post-deposition treatments should be considered as they represent powerful tools to optimize the nano-segregation and orientation of the CNT in the resulting blend/photoactive layer.Performances of CNT electrodes are strongly connected to material qualities such as degree of dispersion, diameter, defect, purity, metallicity and wall number. Equally decisive are interactions between CNT and the other components of the photoactive material, since they impact the optimal energy band offset at the interphase. The fabrication of films with long metallic CNT, which are optimally oriented for charge injection and charge collection at the external circuit, would constitute another milestone toward improved photoelectrodes.
References
- (a) S. Reich, C. Thomsen and J. Maultzsch, Carbon Nanotubes: Basic Concepts and Physical Properties, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed; (b) M. S. Dresselhaus, G. Dresselhaus and P. Avouris, Carbon Nanotubes: Synthesis, Structure, Properties and Applications, Springer, Berlin, Germany, 2001 Search PubMed; (c) P. J. F. Harris, Carbon Nanotubes and Related Structures: New Materials for the Twenty-First Century, Cambridge University Press, Cambridge, UK, 2001 Search PubMed; (d) S. Roth and D. Carroll, One-Dimensional Metals: Conjugated Polymers, Organic Crystals, Carbon Nanotubes, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed; (e) M. Meyyappan, Carbon Nanotubes, Science and Application, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed; (f) Special issue on Carbon Nanotubes, Acc. Chem. Res., 2002, 35, 997 CrossRef; (g) V. Sgobba, G. M. A. Rahman, C. Ehli and D. Guldi, Covalent and Non-covalent Approaches Toward Multifunctional Carbon Nanotubes Materials, in Fullerenes-Principles and Applications, ed. F. Langa and J. F. Nierengarten, RSC, Cambridge, UK, 2007 Search PubMed; (h) V. Sgobba, G. M. A. Rahman and D. M. Guldi, in Carbon Nanotubes in Electron Donor–Acceptor Nanocomposites, Chemistry of Carbon Nanotubes, ed. V. A. Basiuk, American Scientific Publishers, USA, 2006 Search PubMed.
- S. J. Tans, M. H. Devoret, H. Dal, A. Thess, R. E. Smalley, L. J. Geerligs and C. Dekker, Nature, 1997, 386, 474 CrossRef CAS.
- E. Pop, D. Mann, Q. Wang, K. Goodson and H. Dai, Nano Lett., 2006, 6, 96 CrossRef CAS.
- P. M. Ajayan and O. Z. Zhou, Carbon Nanotubes, 2001, 80, 391 Search PubMed.
- J. Robertson, Mater. Today, 2004, 7, 46 CrossRef CAS.
- D. Mann, Carbon Nanotubes, 2006, 19 Search PubMed.
- A. Thess, R. Lee, P. Nikolaev, H. Dai, P. Petit, J. Robert, C. Xu, Y. H. Lee, S. G. Kim, A. G. Rinzler, D. T. Colbert, G. E. Scuseria, D. Tomanek, J. E. Fischer and R. E. Smalley, Science, 1996, 273, 483 CrossRef CAS.
- L. A. Girifalco, M. Hodak and R. S. Lee, Phys. Rev. B: Condens. Matter, 2000, 62, 13104 CrossRef CAS.
- D. Tasis, N. Tagmatarchis, V. Georgakilas and M. Prato, Chem. Eur. J., 2003, 9, 4000 CrossRef CAS.
- M. J. O’Connell, S. M. Bachilo, C. B. Huffman, V. C. Moore, M. S. Strano, E. H. Haroz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kittrell, J. P. Ma, R. H. Hauge, R. B. Weisman and R. E. Smalley, Science, 2002, 297, 593 CrossRef CAS.
- M. Monthioux, B. W. Smith, B. Burteaux, A. Claye, J. E. Fischer and D. E. Luzzi, Carbon, 2001, 39, 1251 CrossRef CAS.
- (a) X. Li, X. Tu, S. Zaric, K. Welsher, W. S. Seo, W. Zhao and H. Dai, J. Am. Chem. Soc., 2007, 129, 15770 CrossRef CAS; (b) B. Wang, L. Wei, L. Yao, L. J. Li, Y. Yang and Y. Chen, J. Phys. Chem. C, 2007, 111, 14612 CrossRef CAS; (c) M. C. Hersam, Nat. Nanotechnol., 2008, 3, 387 Search PubMed.
- (a) M. S. Strano, C. A. Dyke, M. L. Usrey, P. W. Barone, M. J. Allen, H. Shan, C. Kittrell, R. H. Hauge, J. M. Tour and R. E. Smalley, Science, 2003, 301, 1519 CrossRef CAS; (b) G. Zhang, P. Qi, X. Wang, Y. Lu, X. Li, R. Tu, S. Bangsaruntip, D. Mann, L. Zhang and H. Dai, Science, 2006, 314, 974 CrossRef CAS; (c) R. Krupke, F. Hennruch, H. v. Löhneysen and M. M. Kappes, Science, 2003, 301, 344 CrossRef CAS.
- For reviews, see: (a) S. Baneriee, T. Hemraj-Benny and S. S. Wong, Adv. Mater., 2005, 17, 17 CrossRef CAS; (b) A. Hirsch, Angew. Chem., Int. Ed., 2002, 41, 1853 CrossRef CAS; (c) X. Lu and Z. Chen, Chem. Rev., 2005, 105, 3643 CrossRef CAS; (d) J. L. Bahr and J. M. Tour, J. Mater. Chem., 2002, 12, 1952 RSC; (e) D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS.
- P. Nikolaev, M. J. Bronikowski, R. K. Bradley, F. Rohmund, D. T. Colbert, K. A. Smith and R. E. Smalley, Chem. Phys. Lett., 1999, 313, 91 CrossRef CAS.
- (a) K. Mylvaganam and L. C. Zhang, J. Phys. Chem. B, 2004, 108, 5217 CrossRef CAS; (b) X. Lu, F. Tian, X. Xu, N. Wang and Q. Zhang, J. Am. Chem. Soc., 2003, 125, 10459 CrossRef CAS; (c) E. T. Mickelson, C. B. Huffmann, A. G. Rinzler, R. E. Smalley, R. H. Hauge and J. L. Margrave, Chem. Phys. Lett., 1998, 296, 188 CrossRef CAS; (d) H. F. Bettinger, ChemPhysChem, 2003, 4, 1283 CrossRef CAS.
- J. Aihara, J. Phys. Chem. A, 1999, 103, 7487 CrossRef CAS.
- J. Zhao and P. B. Balbuena, J. Phys. Chem. A, 2006, 110, 2771 CrossRef CAS.
- (a) H. Hu, P. Bhowmik, B. Zhao, M. H. Hamon, M. E. Itkis and R. C. Haddon, Chem. Phys. Lett., 2001, 291, 283; (b) A. J. Stone and D. J. Wales, Chem. Phys. Lett., 1986, 128, 501 CrossRef CAS.
- (a) H. F. Bettinger, J. Phys. Chem. B, 2005, 109, 6922 CrossRef CAS; (b) X. Lu, Z. Chen and P. v. R. Schleyer, J. Am. Chem. Soc., 2005, 127, 4313.
- (a) A. Hashimoto, K. Suenaga, A. Gloter, K. Urita and S. Iijima, Nature, 2004, 430, 870 CrossRef CAS; (b) P. C. P. Watts, W. K. Hsu, H. W. Kroto and D. R. M. Walton, Nano Lett., 2003, 333, 549 CrossRef; (c) J. C. Charlier, Acc. Chem. Res., 2002, 35, 1063 CrossRef CAS; (d) M. Ouyang, J. L. Huang, C. L. Cheung and C. M. Lieber, Science, 2001, 291, 97 CrossRef CAS; (e) S. G. Louie, Top. Appl. Phys., 2001, 80, 113 CAS; (f) T. W. Ebbesen and T. Takada, Carbon, 1995, 33, 973 CrossRef CAS; (g) J. C. Charlier, T. W. Ebbesen and P. Lambin, Phys. Rev. B, 1996, 53, 11108 CrossRef CAS; (h) V. H. Crespi and M. L. Cohen, Phys. Rev. Lett., 1997, 79, 2093 CrossRef CAS; (i) L. Chico, M. P. L. Sancho and M. C. Muñoz, Phys. Rev. Lett., 1998, 98, 1278 CrossRef.
- (a) M. Ouyang, J. L. Huang and C. M. Lieber, Acc. Chem. Res., 2002, 35, 1018 CrossRef CAS; (b) Z. Yao, H. W. C. Postma, L. Balents and C. Dekker, Nature, 1999, 402, 273 CrossRef CAS.
- (a) A. F. Collings and C. Critchley, Artificial Photosynthesis: From Basic Biology to Industrial Application, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed; (b) V. Balzani, Electron Transfer in Chemistry, Wiley-VCH, Weinheim, Germany, 2001 Search PubMed; (c) D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22 RSC; (d) J. Deisenhofer and J. R. Norris, The Photosynthetic Reaction Center, Academic Press, San Diego, CA, 1993 Search PubMed; (e) D. M. Guldi, G. M. A. Rahman, V. Sgobba and C. Ehli, Chem. Soc. Rev., 2006, 35, 471 RSC.
- (a) J. Nelson, The Physics of Solar Cells (Properties of Semiconductor Materials), Imperial College Press, London, UK, 2003 Search PubMed; (b) C. Brabec, V. Dyakonov, J. Parisi and S. Sariciftci, Organic Photovoltaics: Concepts and Realization, Springer Series in Materials Science, Springer, Berlin, Germany, 2003 Search PubMed.
- A. Hartschuh, H. N. Pedrosa, J. Peterson, L. Huang, P. Anger, H. Qian, A. J. Meixner, M. Steiner, L. Novotny and T. D. Krauss, ChemPhysChem, 2005, 6, 577 CrossRef CAS.
- N. Grossiord, J. Loos, O. Regev and C. E. Koning, Chem. Mater., 2006, 18, 1089 CrossRef CAS.
- M. Zheng, A. Jagota, M. S. Strano, A. P. Santos, P. Barone, S. G. Chou, B. A. Diner, M. S. Dresselhaus, R. S. Mclean, G. B. Onoa, G. G. Samsonidze, E. D. Semke, M. Usrey and D. J. Walls, Science, 2003, 302, 1545 CrossRef CAS.
- (a) H. Kataura, Y. Kumazawa, Y. Maniwa, I. Umezu, S. Suzuki, Y. Ohtzuka and Y. Achiba, Synth. Met., 1999, 103, 2555 CrossRef CAS; (b) M. A. Amon, M. E. Itkis, S. Niyogi, T. Alvarez, C. Kuper, M. Menon and R. C. Haddon, J. Am. Chem. Soc., 2001, 123, 11292 CrossRef CAS.
- (a) S. Kazaoui, N. Minami, H. K. N. Matsuda and Y. Achiba, Appl. Phys. Lett., 2001, 78, 3433 CrossRef CAS; (b) N. Nair, M. L. Usrey, W. J. Kim, R. D. Braatz and M. S. Strano, Anal. Chem., 2006, 78, 7689 CrossRef CAS.
- (a) V. Perebeinos, J. Tersoff and P. Avouris, Phys. Rev. Lett., 2004, 92, 257402 CrossRef; (b) H. Zhao and S. Mazumdar, Phys. Rev. Lett., 2004, 93, 157402 CrossRef; (c) C. D. Spataru, S. Ismail-Beigi, L. X. Benedict and S. G. Louie, Phys. Rev. Lett., 2004, 92, 077402 CrossRef; (d) Z. Wang, H. Zhao and S. Mazumdar, Phys. Rev. B, 2006, 74, 195406 CrossRef.
- F. Wang, G. Dukovic, L. E. Brus and T. F. Heinz, Science, 2005, 308, 838 CrossRef CAS.
- A. Hartschuh, H. N. Pedrosa, L. Novotny and T. D. Krauss, Science, 2003, 301, 1354 CrossRef CAS.
- S. M. Bachilo, M. S. Strano, C. Kittrell, R. H. Hauge, R. E. Smalley and R. B. Weisman, Science, 2002, 298, 2361 CrossRef CAS.
- M. Jones, C. Engtrakul, W. K. Metzger, R. J. Ellingson, A. J. Nozik, M. J. Heben and G. Rumbles, Phys. Rev. B, 2005, 71, 115426/1 CAS.
- S. M. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Annu. Rev. Phys. Chem., 2007, 58, 719 CrossRef.
- F. Chen, B. Wang, Y. Chen and L. J. Li, Nano Lett., 2007, 7, 3013 CrossRef CAS.
- S. Lebedkin, F. Hennrich, O. Kiowski and M. M. Kappes, Phys. Rev. B, 2008, 77, 165429 CrossRef.
- (a) Y. Zhang, T. Gong, W. Liu, X. Zhang, J. Chang, K. Wang and D. Wu, Appl. Phys. Lett., 2005, 87, 173114 CrossRef; (b) M. E. Brennan, J. N. Coleman, M. Panhuis, L. Marty and W. J. Blau, Synth. Met., 2001, 119, 641 CrossRef CAS.
- F. Wang, G. Dukovic, L. E. Brus and T. F. Heinz, Phys. Rev. Lett., 2004, 92, 177401-1.
- S. B. Cronin, Y. Yin, A. Walsh, R. B. Capaz, A. Stolyarov, P. Tangney, M. L. Cohen, S. G. Louie, A. K. Swan, M. S. Ünlü, B. B. Goldberg and M. Tinkham, Phys. Rev. Lett., 2006, 96, 127403 CrossRef CAS.
- G. Shan and S. Bao, Physica E, 2006, 35, 161 CrossRef CAS.
- (a) A. G. Walsh, A. N. Vamivakas, Y. Yin, S. B. Cronin, M. S. Ünlü, B. B. Goldberg and A. K. Swan, Nano Lett., 2007, 7, 1485 CrossRef CAS; (b) M. T. Woodside and P. L. McEuen, Science, 2002, 296, 1098 CrossRef CAS; (c) P. L. McEuen, M. Bockrath, D. H. Cobden, Y. G. Yoon and S. G. Louie, Phys. Rev. Lett., 1999, 83, 5098 CrossRef CAS.
- S. Zaric, G. N. Ostojic, J. Kono, J. Shaver, V. C. Moore, M. S. Strano, R. H. Hauge, R. E. Smalley and X. Wei, Science, 2004, 304, 1129 CrossRef CAS.
- (a) P. W. Barone, R. S. Parker and M. S. Strano, Anal. Chem., 2005, 77, 7556 CrossRef CAS; (b) P. W. Barone, S. Baik, D. A. Heller and M. S. Strano, Nat. Mater., 2005, 4, 86 CrossRef CAS.
- J. A. Misevich, R. Martel, P. Avouris, J. C. Tsang, S. Heinze and J. Tersoff, Science, 2003, 300, 783 CrossRef CAS.
- S. Kazaoui, N. Minami, R. Jacquemin, H. Kataura and Y. Achiba, Phys. Rev. B, 1999, 60, 13339 CrossRef CAS.
- (a) P. Petit, C. Mathis, C. Journet and P. Bernier, Chem. Phys. Lett., 1999, 305, 370 CrossRef CAS; (b) E. Jouguelet, C. Mathis and P. Petit, Chem. Phys. Lett., 2000, 318, 561 CrossRef CAS.
- A. M. Rao, P. C. Eklund, S. Bandow, A. Thess and R. E. Smalley, Nature, 1997, 388, 257 CrossRef CAS.
- L. Kavan and L. Dunsch, ChemPhysChem, 2007, 8, 974 CrossRef CAS.
- R. Jacquemin, S. Kazaoui, D. Yu, A. Hassanien, N. Minami, H. Kataura and Y. Achiba, Synth. Met., 2000, 115, 283 CrossRef CAS.
- N. Bendiab, E. Anglaret, J. L. Bantignies, A. Zahab, J. L. Sauvajol, P. Petit and C. Mathis, Phys. Rev. B, 2001, 64, 245424 CrossRef.
- H. Son, A. Reina, G. G. Samsonidze, R. Saito, A. Jorio, M. S. Dresselhaus and J. Kong, Phys. Rev. B, 2006, 74, 073406 CrossRef.
- (a) L. Kavan, P. Rapta, L. Dunsch, M. J. Bronikowski, P. Willis and R. E. Smalley, J. Phys. Chem. B, 2001, 105, 10764 CrossRef CAS; (b) S. Gupta, M. Hughes, A. H. Windle and J. Robertson, J. Appl. Phys., 2004, 95, 2038 CrossRef CAS.
- L. Kavan, M. Kalbáč, M. Zukalová and L. Dunsch, J. Phys. Chem. B, 2005, 109, 19613 CrossRef CAS.
- P. M. Rafailov, J. Maultzsch, C. Thomsen and H. Kataura, Phys. Rev. B, 2005, 72, 045411 CrossRef.
- M. S. Dresselhaus and G. Dresselhaus, Adv. Phys., 1981, 30, 139 CAS.
- (a) A. Claye, S. Rahman, J. E. Fischer, A. Sirenko, G. U. Sumanasekera and P. C. Eklund, Chem. Phys. Lett., 2001, 333, 16 CrossRef CAS; (b) M. Stoll, P. M. Rafailov, W. Frenzel and C. Thomsen, Chem. Phys. Lett., 2003, 375, 625 CrossRef CAS; (c) S. Gupta and J. Robertson, J. Appl. Phys., 2006, 100, 083711 CrossRef.
- (a) L. Kavan, P. Rapta and L. Dunsch, Chem. Phys. Lett., 2000, 328, 363 CrossRef CAS; (b) P. Corio, P. S. Santos, V. W. Brar, G. G. Samsonidze, S. G. Chou and M. S. Dresselhaus, Chem. Phys. Lett., 2003, 370, 675 CrossRef CAS; (c) L. Kavan and L. Dunsch, Nano Lett., 2003, 3, 969 CrossRef CAS; (d) P. Corio, A. Jorio, N. Demir and M. S. Dresselhaus, Chem. Phys. Lett., 2004, 392, 396 CrossRef CAS; (e) L. Kavan and L. Dunsch, ChemPhysChem, 2003, 4, 944 CrossRef; (f) K. Murakoshi and K. Okazaki, Electrochim. Acta, 2005, 50, 3069 CrossRef CAS.
- K. Okazaki, Y. Nakato and K. Murakoshi, Phys. Rev. B, 2003, 68, 035434 CrossRef.
- J. Cambedouzou, J. L. Sauvajol, A. Rahmani, E. Flahaut, A. Peigney and C. Laurent, Phys. Rev. B, 2005, 69, 235422.
- A. Pénicaud, P. Poulin, A. Derré, E. Anglaret and P. Petit, J. Am. Chem. Soc., 2005, 127, 8 CrossRef CAS.
- D. Paolucci, M. M. Franco, M. Iurlo, M. Marcaccio, M. Prato, F. Zerbetto, A. Pénicaud and F. Paolucci, J. Am. Chem. Soc., 2008, 130, 7393 CrossRef CAS.
- Z. Wang, H. Pedrosa, T. Krauss and L. Rothberg, Phys. Rev. Lett., 2007, 98, 019702 CrossRef.
- P. M. Ajayan, T. W. Ebbesen, T. Ichihashi, S. Iijima, K. Tanigaki and H. Hiura, Nature, 1993, 362, 522 CrossRef CAS.
- (a) J. Fan, M. Yudasaka, J. Miyawaki, K. Ajima, K. Murata and S. Iijima, J. Phys. Chem. B, 2006, 110, 1587 CrossRef CAS; (b) C. A. Furtado, U. J. Kim, H. R. Gutierrez, L. Pan, E. C. Dickey and P. C. Eklund, J. Am. Chem. Soc., 2004, 126, 6095151; (c) R. Sen, S. M. Rickard, M. E. Itkis and R. C. Haddon, Chem. Mater., 2003, 15, 4273 CrossRef CAS.
- E. Dujardin, T. W. Ebbesen, A. Treacy and M. M. J. Krishnan, Adv. Mater., 1998, 10, 611 CrossRef CAS.
- P. M. Ajayan and S. Iijima, Nature, 1993, 361, 333 CrossRef CAS.
- (a) M. Shim, J. H. Back, T. Ozel and K. W. Kwon, Phys. Rev. B, 2005, 71, 205411/1 CAS; (b) Y. Miyamoto, N. Jinbo, H. Nakamura, A. Rubio and D. Tomanek, Phys. Rev. B, 2004, 70, 233408 CrossRef; (c) P. G. Collins, K. Bradley, M. Ishigami and A. Zettl, Science, 2000, 287, 1801 CrossRef CAS.
- (a) R. K. Saini, I. W. Chiang, H. Peng, R. E. Smalley, W. E. Billups, R. H. Hauge and J. L. Margrave, J. Am. Chem. Soc., 2003, 125, 3617 CrossRef CAS; (b) P. J. Boul, J. Liu, E. T. Mickelson, C. B. Huffman, L. M. Ericson, I. W. Chiang, K. A. Smith, D. T. Colbert, R. H. Hauge, J. L. Margrave and R. E. Smalley, Chem. Phys. Lett., 1999, 310, 367 CrossRef CAS.
- T. Kawai and Y. Miyamoto, Chem. Phys. Lett., 2008, 453, 256 CrossRef CAS.
- D. B. Mawhinney, V. Naumenko, A. Kuznetsova, J. T. Yates Jr, J. Liu and R. E. Smalley, Chem. Phys. Lett., 2000, 324, 213 CrossRef.
- J. Chen, A. M. Rao, S. Lyuksyutov, M. E. Itkis, M. A. Hamon, H. Hu, R. W. Cohn, P. C. Eklund, D. T. Colbert, R. E. Smalley and R. C. Haddon, J. Phys. Chem. B, 2001, 105, 2525 CrossRef CAS.
- H. Hu, P. Bhowmik, B. Zhao, M. A. Hamon, M. E. Itkis and R. C. Haddon, Chem. Phys. Lett., 2001, 345, 25 CrossRef CAS.
- X. Li, J. Niu, J. Zhang, H. Li and Z. Liu, J. Phys. Chem. B, 2003, 107, 2453 CrossRef CAS.
- W. Wand, P. Serp, P. Kalck and J. L. Faria, J. Mol. Catal., 2005, 235, 294.
- L. Pietronero and S. Strassler, Phys. Rev. Lett., 1981, 47, 593 CrossRef CAS.
- M. J. O’Connell, E. E. Eibergen and S. K. Doorn, Nat. Mater., 2005, 4, 412 CrossRef CAS.
- W. Zhou, J. Vavro, N. M. Nemes, J. E. Fischer, F. Borondics, K. Kamaras and D. B. Tanner, Phys. Rev. B, 2005, 71, 205423 CrossRef.
- R. Graupner, J. Abraham, A. Vencelová, T. Seyller, F. Hennrich, M. M. Kappes, A. Hirsch and L. Ley, Phys. Chem. Chem. Phys., 2003, 5, 5472 RSC.
- (a) G. U. Sumanasekera, C. K. W. Adu, S. Fang and P. C. Eklund, Phys. Rev. Lett., 2000, 85, 1096 CrossRef CAS; (b) P. J. Blatt, P. A. Schroeder, C. L. Foiles and D. Greig, in Thermoelectric Power of Metals, Plenum, New York and London, 1976, ch. 2 Search PubMed.
- S. J. Tans, A. R. M. Verschueren and C. Dekker, Nature, 1998, 393, 49 CrossRef CAS.
- C. Zhou, J. Kong, E. Yenilmez and H. Dai, Science, 2000, 290, 1552 CrossRef CAS.
- S. B. Fagan, A. G. Souza Filho, J. Mendes Filho, P. Corio and M. S. Dresselhaus, Chem. Phys. Lett., 2005, 456, 54 CrossRef.
- P. Corio, A. P. Santos, M. L. A. Temperini, V. W. Brar, M. A. Pimenta and M. S. Dresselhaus, Chem. Phys. Lett., 2004, 383, 475 CrossRef CAS.
- B. R. Sankapal, K. Setyowati, J. Chen and H. Liu, Appl. Phys. Lett., 2007, 91, 173103/1 CAS.
- (a) V. Z. Mordkovitch, M. Baxendale, S. Yoshimura and R. P. H. Chang, Carbon, 1996, 34, 1301 CrossRef; (b) M. Baxendale, V. Z. Mordkovich, S. Yoshimura, R. P. H. Chang and A. G. M. Jansen, Phys. Rev. B, 1998, 57, 15629 CrossRef CAS.
- (a) D. Claves, J. Giraudet, M. C. Schouler, P. Gadelle and A. Hamwi, Solid State Commun., 2004, 130, 1 CrossRef CAS; (b) V. Z. Mordkovitch, M. Baxendale, R. P. Chang and H. Yoshimura, Synth. Met., 1997, 86, 2049 CrossRef CAS; (c) V. Z. Mordkovitch, Mol. Cryst. Liq. Cryst., 2000, 340, 775 CrossRef.
- V. Skákalová, A. B. Kaiser, U. Dettlaff-Wegligowska, K. Hrnčariková and S. Roth, J. Phys. Chem. B, 2005, 109, 7174 CrossRef CAS.
- M. S. Strano, C. B. Huffman, V. C. Moore, M. J. O'Connell, E. H. Haroz, J. Hubbard, M. Miller, K. Rialon, C. Kittrell, S. Ramesh, R. H. Hauge and R. E. Smalley, J. Phys. Chem. B, 2003, 107, 6979 CrossRef CAS.
- L. Eberson and F. Radner, Acta Chem. Scand., 1991, 45, 1093 CAS.
- C. Klinke, A. Afzali and P. Avouris, Chem. Phys. Lett., 2006, 430, 75 CrossRef CAS.
- P. Gai, O. Stephan, K. McGuire, A. M. Rao, M. S. Dresselhaus, G. Dresselhaus and C. Colliex, J. Mater. Chem., 2004, 14, 669 RSC.
- E. Borowiak-Palen, T. Pichler, G. G. Fuentes, A. Graff, R. J. Kalenczuk, M. Knupfer and J. Fink, Chem. Phys. Lett., 2003, 378, 516 CrossRef CAS.
- M. Terrones, W. K. Hsu, A. Schilder, H. Terrones, N. Grobert, J. P. Hare, Y. Q. Zhu, M. Schwoerer, K. Prassides, H. W. Kroto and D. R. M. Walton, Appl. Phys. A: Mater., 1998, 66, 307 CrossRef.
- R. Czerw, M. Terrones, J. C. Charlier, X. Blase, B. Foley, R. Kamalakaran, N. Grobert, H. Terrones, P. M. Ajayan, W. Blau, D. Tekleab, M. Rühle and D. L. Carroll, Nano Lett., 2001, 1, 457 CrossRef CAS.
- M. Glerup, J. Steinmetz, D. Samaille, O. Stephan, S. Enouz, A. Loiseau, S. Roth and P. Bernier, Chem. Phys. Lett., 2004, 387, 193 CrossRef CAS.
- R. Sen, B. C. Satishkumar, S. Govindaraj, K. R. Harikumar, M. K. Renganathan and C. N. R. Rao, J. Mater. Chem., 1997, 7, 2335 RSC.
- M. Terrones, N. Grobert, J. Olivares, J. P. Zhang, H. Terrones, K. Kordatos, W. K. Hsu, J. P. Hare, P. D. Townsend, K. Prassides, A. K. Cheetham, H. W. Kroto and D. R. M. Walton, Nature, 1997, 388, 52 CrossRef CAS.
- F. Villalpando-Paez, A. Zamudio, A. L. Elias, H. Son, E. B. Barros, S. Chou, Y. A. Kim, H. Muramatsu, T. Hayashi, J. Kong, H. Terrones, G. Dresselhaus, M. Endo, M. Terrones and M. S. Dresselhaus, Chem. Phys. Lett., 2006, 424, 345 CrossRef CAS.
- D. Golberg, Y. Bando, L. Bourgeois, K. Kurashima and T. Sato, Carbon, 2000, 38, 2017 CrossRef CAS.
- (a) O. Stephan, P. M. Ajayan, C. Colliex, P. Redlich, J. M. Lambert, P. Bernier and P. Lefin, Science, 1994, 266, 1683 CAS; (b) K. Suenaga, C. Colliex, N. Demoncy, A. Loiseau, H. Pascard and F. Willaime, Science, 1997, 278, 653 CrossRef CAS.
- M. Terrones, A. M. Benito, C. Manteca-Diego, W. K. Hsu, O. I. Osman, J. P. Hare, D. G. Reid, H. Terrones, A. K. Cheetham, K. Prassides, H. W. Kroto and D. R. M. Walton, Chem. Phys. Lett., 1996, 257, 576 CrossRef CAS.
- R. J. Baierle, S. B. Fagan, R. Mota, A. J. R. da Silva and A. Fazzio, Phys. Rev., 2001, 64, 085413.
- E. Cruz-Silva, D. Cullen, L. Gu, J. M. Romo-Herrera, E. Múnoz-Sandoval, F. López-Urías, D. J. Smith, H. Terrones and M. Terrones, ACS Nano, 2008, 2, 441 CrossRef CAS.
- (a) M. Monthioux, Carbon, 2002, 40, 1809 CrossRef CAS; (b) A. N. Khlobystov, D. A. Britz and G. A. D. Briggs, Acc. Chem. Res., 2005, 38, 901 CrossRef CAS; (c) O. Vostrowsky and A. Hirsch, Angew. Chem., Int. Ed., 2004, 43, 2326 CrossRef CAS; (d) J. loan, A. I. Kirkland, J. L. Hutchison and M. L. H. Green, Acc. Chem. Res., 2002, 35, 1054 CrossRef CAS.
- P. W. Chiu, S. F. Yang, S. H. Yang, G. Gu and S. Roth, Appl. Phys. A, 2003, 76, 463 CrossRef CAS.
- J. Lee, H. Kim, S. J. Kahng, G. Kim, Y. W. Son, J. Ihm, H. Kato, Z. W. Wang, T. Okazaki, H. Shinohara and Y. Kuk, Nature, 2002, 415, 1005 CrossRef CAS.
- S. Okada, S. Saito and A. Oshiyama, Phys. Rev. Lett., 2001, 86, 3835 CrossRef CAS.
- J. Vavro, M. C. Llaguno, B. C. Satishkumar, D. E. Luzzi and J. E. Fischer, Appl. Phys. Lett., 2002, 80, 1450 CrossRef CAS.
- D. J. Hornbaker, S. J. Kahng, S. Misra, B. W. Smith, A. T. Johnson, E. J. Mele, D. E. Luzzi and A. Yazdani, Science, 2002, 295, 828 CrossRef CAS.
- Y. Cho, S. Han, G. Kim, H. Lee and J. Ihm, Phys. Rev. Lett., 2003, 90, 106402/1 CAS.
- A. Debarre, R. Julien, D. Nutarelli, A. Richard and P. Tchenio, Phys. Rev. Lett., 2003, 91, 085501/1 CAS.
- L. J. Li, A. N. Khlobystov, J. G. Wiltshire, G. A. Briggs and R. J. Nicholas, Nat. Mater., 2005, 4, 534 CrossRef CAS.
- T. Takenobu, T. Takano, M. Shiraishi, Y. Murakami, M. Ata, H. Kataura, Y. Achiba and Y. Iwasa, Nat. Mater., 2003, 2, 683 CrossRef CAS.
- L. Duclaux, J. P. Salvetat, P. Lauginie, T. Cacciaguera, A. M. Faugere, C. Goze-Bac and P. Bernier, J. Phys. Chem. Solids, 2003, 64, 571 CrossRef CAS.
- K. Bradley, S. H. Jhi, P. G. Collins, J. Hone, M. L. Cohen, S. G. Louie and A. Zettl, Phys. Rev. Lett., 2000, 85, 4361 CrossRef CAS.
- (a) V. Derycke, R. Martel, J. Appenzeller and P. Avouris, Nano Lett., 2001, 1, 453 CrossRef CAS; (b) X. Liu, C. Lee, C. Zhou and J. Han, Appl. Phys. Lett., 2001, 79, 3329 CrossRef CAS.
- R. Martel, V. Derycke, C. Lavoie, J. Appenzeller, K. Chan, J. Tersoff and P. Avouris, Phys. Rev. Lett., 2001, 87, 256805 CrossRef CAS.
- D. Kang, N. Park, J. Ko, E. Bae and W. Park, Nanotechnology, 2005, 16, 1048 CrossRef CAS.
- S. H. Jhi, S. G. Louie and M. L. Cohen, Phys. Rev. Lett., 2000, 85, 1710 CrossRef CAS.
- J. Kong, N. R. Franklin, C. Zhou, M. G. Chapline, S. Peng, K. Cho and H. Dai, Science, 2000, 287, 622 CrossRef CAS.
- M. J. O’Connell, E. E. Eibergen and S. K. Doorn, Nat. Mater., 2005, 4, 412 CrossRef CAS.
- J. Oh, S. Roh, W. Yi, H. Lee and J. Yoo, J. Vac. Sci. Technol., B, 2004, 22, 1416 CrossRef CAS.
- M. Shim, T. Ozel, A. Gaur and C. Wang, J. Am. Chem. Soc., 2006, 128, 7522 CrossRef CAS.
- H. Z. Geng, K. K. Kim, C. Song, N. T. Xuyen, S. M. Kim, K. A. Park, D. S. Lee, K. H. An, Y. S. Lee, Y. Chang, Y. J. Lee, J. Y. Choi, A. Benayad and Y. H. Lee, J. Mater. Chem., 2008, 18, 1261 RSC.
- J. Kong and H. Dai, J. Phys. Chem. B, 2001, 105, 2890 CrossRef CAS.
- D. M. Guldi, G. M. A. Rahman, F. Zerbetto and M. Prato, Acc. Chem. Res., 2005, 38, 871 CrossRef CAS.
- (a) K. Kalyanasundaram, Photochemistry of Polypyridine and Porphyrin Complexes, Academic Press, London, 1997 Search PubMed; (b) D. Dolphin, The Porphyrins, Academic Press, New York, 1978 Search PubMed; (c) K. M. Kadish, K. M. Smith and R. Guilard, The Porphyrin Handbook, Academic Press, New York, USA, 2003 Search PubMed.
- (a) D. Baskaran, J. W. Mays, X. P. Zhang and M. S. Bratcher, J. Am. Chem. Soc., 2005, 127, 6916 CrossRef CAS; (b) H. Li, R. B. Martin, B. A. Harruff, R. A. Carino, L. F. Allard and Y. P. Sun, Adv. Mater., 2004, 16, 896 CrossRef CAS.
- (a) C. C. Leznoff and A. B. P. Lever, Phthalocyanines: Properties and Applications, VCH, Weinheim, Germany, 1989, 1993, 1996 Search PubMed; (b) N. B. McKeown, Phthalocyanine Materials: Synthesis, Structure and Function, Cambridge University Press, Cambridge, UK, 1998 Search PubMed; (c) G. de la Torre, P. Vázquez, F. Agulló-López and T. Torres, Chem. Rev., 2004, 104, 3723 CrossRef CAS.
- (a) G. de la Torre, W. Blau and T. Torres, Nanotechnology, 2003, 14, 765 CrossRef CAS; (b) H. B. Xu, H. Z. Chen, M. M. Shi, R. Bai and M. Wang, Mater. Chem. Phys., 2005, 94, 342 CrossRef CAS.
- (a) J. L. Segura and N. Martin, Angew. Chem., Int. Ed., 2001, 40, 1372 CrossRef CAS; (b) N. Martin, L. Sanchez, M. A. Herranz, B. Illescas and D. M. Guldi, Acc. Chem. Res., 2007, 40, 1015 CrossRef CAS.
- M. Á. Herranz, N. Martin, S. Campidelli, M. Prato, G. Brehm and D. M. Guldi, Angew. Chem., Int. Ed., 2006, 45, 4478 CrossRef CAS.
- N. W. S. Kam, T. C. Jessop, P. A. Wender and H. Dai, J. Am. Chem. Soc., 2004, 126, 6850 CrossRef CAS.
- (a) T. Y. Lee and J. B. Yoo, Diamond Rel. Mater., 2005, 14, 1888 CrossRef CAS; (b) S. R. Jang, R. Vittal and K. J. Kim, Langmuir, 2004, 20, 9807 CrossRef CAS.
- B. O’Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef CAS.
- (a) R. F. Khairoutdinov, L. V. Doubova, R. C. Haddon and L. Saraf, J. Phys. Chem. B, 2004, 108, 19976 CrossRef CAS; (b) F. Frehill, J. G. Vos, S. Benrezzak, A. A. Koos, Z. Konya, M. G. Ruether, W. J. Blau, A. Fonseca, J. B. Nagy, L. P. Biro, A. I. Minett and M. in het Panhuis, J. Am. Chem. Soc., 2002, 124, 13694 CrossRef CAS.
- P. Wang, C. N. Moorefield, S. Li, S. H. Hwang, C. D. Shreiner and G. R. Newkome, Chem. Commun., 2006, 1091 RSC.
- J. Liu, A. G. Rinzler, H. Dai, J. H. Hafner, R. K. Bradley, P. J. Boul, A. Lu, T. Iverson, K. Shelimov, C. B. Huffman, F. Rodrigues-Macias, Y. S. Shon, T. R. Lee, D. T. Colbert and R. E. Smalley, Science, 1998, 280, 1253 CrossRef CAS.
- A. Zamudio, A. L. Elías, J. A. Rodríguez-Manzo, F. López-Urías, G. Rodríguez-Gattorno, F. Lupo, M. Rühle, D. J. Smith, H. Terrones, D. Díaz and M. Terrones, Small, 2006, 2, 346 CrossRef CAS.
- R. Zanella, E. Basiuk, P. Santiago, V. A. Basiuk, Edgar Mireles, I. Puente-Lee and J. M. Saniger, J. Phys. Chem. B, 2005, 109, 16290 CrossRef CAS.
- L. Sheeney-Haj-Ichia, B. Basnar and I. Willner, Angew. Chem., Int. Ed., 2005, 44, 78 CrossRef.
- B. Pan, D. Cui, R. He, F. Gao and Y. Zhang, Chem. Phys. Lett., 2006, 417, 419 CrossRef CAS.
- S. Banerjee and S. S. Wong, Nano Lett., 2002, 2, 195 CrossRef CAS.
- S. Ravindran, S. Chaudhary, B. Colburn, M. Ozkan and C. S. Ozkan, Nano Lett., 2003, 3, 447 CrossRef CAS.
- (a) P. Diao and Z. Liu, J. Phys. Chem. B, 2005, 109, 20906 CrossRef CAS; (b) L. Cai, J. L. Bahr, Y. Yao and J. M. Tour, Chem. Mater., 2002, 14, 4235 CrossRef CAS; (c) P. Diao, Z. Liu, B. Wu, X. Nan, J. Zhang and Z. Wei, ChemPhysChem, 2002, 12, 898 CrossRef.
- (a) M. Burghard, Small, 2005, 1, 1148 CrossRef CAS; (b) K. M. Lee, L. Li and L. Dai, J. Am. Chem. Soc., 2005, 127, 4122 CrossRef CAS; (c) N. Chopra, M. Majumder and B. J. Hinds, Adv. Funct. Mater., 2005, 15, 858 CrossRef CAS.
- Z. Wei, M. Kondratenko, L. H. Dao and D. M. Perepichka, J. Am. Chem. Soc., 2006, 128, 3134 CrossRef CAS.
- D. M. Guldi, M. Marcaccio, D. Paolucci, F. Paolucci, N. Tagmatarchis, D. Tasis, E. Vázquez and M. Prato, Angew. Chem., Int. Ed., 2003, 42, 4206 CrossRef.
- S. Campidelli, C. Sooambar, E. L. Diz, C. Ehli, D. M. Guldi and M. Prato, J. Am. Chem. Soc., 2006, 128, 12544 CrossRef CAS.
- B. Ballesteros, S. Campidelli, G. de la Torre, C. Ehli, D. M. Guldi, M. Prato and T. Torres, Chem. Commun., 2007, 2950 RSC.
- B. Ballesteros, G. de la Torre, C. Ehli, G. M. A. Rahman, F. Agullo-Rueda, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2007, 129, 5061 CrossRef CAS.
- M. Alvaro, P. Atienzar, P. de la Cruz, J. L. Delgardo, V. Troiani, H. Garcia, F. Langa, A. Palkar and L. Echegoyen, J. Am. Chem. Soc., 2006, 128, 6626 CrossRef CAS.
- J. K. Lim, B. K. Yoo, W. Yi, S. Hong, H. Y. Paik, K. Chun, S. K. Kim and S. W. Joo, J. Mater. Chem., 2006, 16, 2374 RSC.
- Z. Guo, F. Du, D. Ren, Y. Chen, J. Zheng, Z. Liu and J. Tian, J. Mater. Chem., 2006, 16, 3021 RSC.
- (a) S. Qin, D. Qin, W. T. Ford, J. E. Herrera, D. E. Resasco, S. M. Bachilo and R. B. Weisman, Macromolecules, 2004, 35, 3965 CrossRef CAS; (b) D. M. Guldi, G. M. A. Rahman, J. Ramey, M. Marcaccio, D. Paolucci, F. Paolucci, S. Qin, W. T. Ford, D. Balbinot, N. Jux, N. Tagmatarchis and M. Prato, Chem. Commun., 2004, 2034 RSC.
- D. M. Guldi, G. M. A. Rahman, S. Qin, M. Tchoul, W. T. Ford, M. Marcaccio, D. Paolucci, F. Paolucci, S. Campidelli and M. Prato, Chem. Eur. J., 2006, 12, 2152 CrossRef CAS.
- G. M. A. Rahman, A. Troeger, V. Sgobba, D. M. Guldi, N. Jux, M. N. Tchoul, W. T. Ford, A. Mateo-Alonso and M. Prato, Chem. Eur. J., 2008, 14, 8837 CrossRef CAS.
- N. Nakashima, Y. Tomonari and H. Murakami, Chem. Lett., 2002, 638 CrossRef CAS.
- H. Paloniemi, T. Ääritalo, T. Laiho, H. Luike, N. Kocharova, K. Haapakka, F. Terzi, R. Seeber and J. Lukkari, J. Phys. Chem. B, 2005, 109, 8634 CrossRef CAS.
- D. M. Guldi, G. M. A. Rahman, N. Jux, N. Tagmatarchis and M. Prato, Angew. Chem., Int. Ed., 2004, 43, 5526 CrossRef CAS.
- C. Ehli, G. M. A. Rahman, N. Jux, D. Balbinot, D. M. Guldi, F. Paolucci, F. Marcaccio, D. Paolucci, M. Melle-Franco, F. Zerbetto, S. Campidelli and M. Prato, J. Am. Chem. Soc., 2006, 128, 11222 CrossRef CAS.
- D. M. Guldi, E. Menna, M. Maggini, M. Marcaccio, D. Paolucci, F. Paolucci, S. Campidelli, M. Prato, G. M. A. Rahman and S. Schergna, Chem. Eur. J., 2006, 12, 3975 CrossRef CAS.
- D. M. Guldi, G. M. A. Rahman, N. Jux, N. Tagmatarchis and M. Prato, Angew. Chem., Int. Ed., 2004, 43, 5526 CrossRef CAS.
- G. M. A. Rahman, D. M. Guldi, R. Cagnoli, A. Mucci, L. Schenetti, L. Vaccari and M. Prato, J. Am. Chem. Soc., 2005, 127, 10051 CrossRef CAS.
- D. M. Guldi, G. M. A. Rahman, V. Sgobba, N. A. Kotov, D. Bonifazi and M. Prato, J. Am. Chem. Soc., 2006, 128, 2315 CrossRef CAS.
- A. Satake, Y. Miyajima and Y. Kobuke, Chem. Mater., 2005, 17, 716 CrossRef CAS.
- F. Cheng and A. Adronov, Chem. Eur. J., 2006, 12, 5053 CrossRef CAS.
- F. Cheng, S. Zhang, A. Adronov, L. Echegoyen and F. Diederich, Chem. Eur. J., 2006, 12, 6062 CrossRef CAS.
- H. Murakami, T. Nomura and N. Nakashima, Chem. Phys. Lett., 2003, 378, 481 CrossRef CAS.
- G. M. A. Rahman, D. M. Guldi, S. Campidelli and M. Prato, J. Mater. Chem., 2006, 16, 62 RSC.
- J. Chen and C. P. Collier, J. Phys. Chem. B, 2005, 109, 7605 CrossRef CAS.
- H. Li, B. Zhou, Y. Lin, L. Gu, W. Wang, K. A. S. Fernando, S. Kumar, L. F. Allard and Y. P. Sun, J. Am. Chem. Soc., 2004, 126, 1014 CrossRef CAS.
- D. S. Hecht, R. J. A. Ramirez, M. Briman, E. Artukovic, K. S. Chichak, J. F. Stoddart and G. Gruener, Nano Lett., 2006, 6, 2031 CrossRef CAS.
- F. D’Souza, R. Chitta, A. S. D. Sandanayaka, N. K. Subbaiyan, L. D'Souza, Y. Araki and O. Ito, J. Am. Chem. Soc., 2007, 129, 15865 CrossRef CAS.
- P. J. Boul, D. G. Cho, G. M. A. Rahman, M. Marquez, Z. Ou, K. M. Kadish, D. M. Guldi and J. L. Sessler, J. Am. Chem. Soc., 2007, 129, 5683 CrossRef CAS.
- (a) G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CrossRef CAS; (b) M. Granstrom, K. Petritsch, A. C. Arias, A. Lux, M. R. Andersson and R. H. Friend, Nature, 1998, 395, 257 CrossRef CAS.
- (a) D. H. Lien, H. W. Zan, N. H. Tai and C. H. Tsai, Adv. Mater., 2006, 18, 98 CrossRef CAS; (b) J. L. Sun, J. Wei, J. L. Zhu, D. Xu, X. Liu, H. Sun, D. H. Wu and N. L. Wu, Appl. Phys. Lett., 2006, 88, 131107 CrossRef.
- M. Cinke, J. Li, B. Chen, A. Cassell, L. Delzeit, J. Han and M. Meyyappan, Chem. Phys. Lett., 2002, 365, 69 CrossRef CAS.
- V. Sgobba and D. M. Guldi, J. Mater. Chem., 2008, 18, 153 RSC.
- D. Romero, M. Carrard, W. De Heer and L. Zuppiroli, Adv. Mater., 1996, 8, 899 CAS.
- (a) E. Kymakis and G. A. J. Amaratunga, Appl. Phys. Lett., 2002, 80, 112 CrossRef CAS; (b) E. Kymakis, I. Alexandrou and G. A. J. Amaratunga, J. Appl. Phys., 2003, 93, 1764 CrossRef CAS; (c) E. Kymakis and G. A. J. Amaratunga, Sol. Energy Mater. Sol. Cells, 2003, 80, 465 CrossRef CAS; (d) J. Landi, R. P. Raffaelle, S. L. Castro and S. G. Bailey, Prog. Photovoltaics, 2005, 13, 165 CAS; (e) Z. Xu, Y. Wu, B. Hu, I. N. Ivanov and D. B. Geohegan, Appl. Phys. Lett., 2005, 87, 263118 CrossRef.
- (a) A. J. Miller, R. A. Hatton, P. Ravi and S. Silva, Appl. Phys. Lett., 2006, 89, 123115/1 CAS; (b) A. J. Miller, R. A. Hatton, S. Silva and P. Ravi, Appl. Phys. Lett., 2006, 89, 133117/1 CAS.
- (a) B. Pradhan, S. K. Batabyal and A. J. Pal, Appl. Phys. Lett., 2006, 88, 093106 CrossRef; (b) C. Li, Y. Chen, Y. Wang, Z. Iqbal, M. Chhowalla and S. Mitra, J. Mater. Chem., 2007, 17, 2406 RSC; (c) G. Kalita, S. Adhikari, H. R. Aryal, M. Umeno, R. Afre, T. Soga and M. Sharon, Appl. Phys. Lett., 2008, 92, 063508 CrossRef.
- (a) B. J. Landi, S. L. Castro, H. J. Ruf, C. M. Evans, S. G. Bailey and R. P. Raffaelle, Sol. Energy Mater. Sol. Cells, 2005, 87, 733 CrossRef CAS; (b) J. A. Rud, L. S. Lovell, J. W. Senn, Q. Qiao and J. T. McLeskey, Jr, J. Mater. Sci., 2005, 40, 1455 CrossRef CAS.
- Y. Kim, S. A. Choulis, J. Nelson, D. D. C. Bradley, S. Cook and J. R. Durrant, Appl. Phys. Lett., 2005, 86, 6272.
- J. J. Dittmer, E. A. Marseglia and R. H. Friend, Adv. Mater., 2000, 12, 1270 CrossRef CAS.
- E. Kymakis, E. Koudoumas, I. Franghiadakis and G. A. J. Amaratunga, J. Phys. D: Appl. Phys., 2006, 39, 1058 CrossRef CAS.
- S. Kazaoui, N. Minami, B. Nalini, Y. Kim and K. Hara, J. Appl. Phys., 2005, 98, 084314 CrossRef.
- J. Geng and T. Zeng, J. Am. Chem. Soc., 2006, 128, 16827 CrossRef CAS.
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864 CAS.
- Y. Kanai and J. C. Grossman, Nano Lett., 2008, 8, 908 CrossRef CAS.
- E. Kymakis and G. A. J. Amaratunga, Rev. Adv. Mater. Sci., 2005, 8, 300.
- C. D. Canestraro, M. C. Scnitzler, A. J. G. Zarbin, M. G. E. da Luz and L. S. Roman, Appl. Surf. Sci., 2006, 252, 5575 CrossRef CAS.
- S. Chaudhary, H. Lu, A. M. Müller, C. J. Bardeen and M. Ozkan, Nano Lett., 2007, 7, 1973 CrossRef CAS.
- (a) D. M. Guldi, G. M. A. Rahman, M. Prato, N. Jux, S. Qin and W. T. Ford, Angew. Chem., Int. Ed., 2005, 44, 2015 CrossRef CAS; (b) D. M. Guldi, G. M. A. Rahman, V. Sgobba, S. Campidelli and M. Prato, Adv. Mater., 2006, 18, 2264 CrossRef.
- (a) V. Sgobba, C. Schulz-Drost and D. M. Guldi, Chem. Commun., 2007, 565 RSC; (b) D. M. Guldi, I. Zilbermann, G. Anderson, N. A. Kotov, N. Tagmatarchis and M. Prato, J. Am. Chem. Soc., 2004, 126, 14340 CrossRef CAS; (c) V. Sgobba, C. Schulz-Drost and D. M. Guldi, Chem. Commun., 2007, 6 Search PubMed; (d) C. Schulz-Drost, V. Sgobba and D. M. Guldi, J. Phys. Chem. C, 2007, 111, 9694 CrossRef CAS; (e) C. Gerhards, C. Schulz-Drost, V. Sgobba and D. M. Guldi, J. Phys. Chem. B, 2008 DOI:10.1021/jp8030094.
- F. Itoh, I. Suzuki and K. Miyairi, Jpn. J. Appl. Chem., 2005, 44, 636 Search PubMed.
- (a) S. Barazzouk, S. Hotchandani, K. Vinodgopal and P. V. Kamat, J. Phys. Chem. B, 2004, 108, 17015 CrossRef CAS; (b) P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834 CrossRef CAS.
- (a) I. Robel, B. A. Bunker and P. V. Kamat, Adv. Mater., 2005, 17, 2458 CrossRef CAS; (b) T. Hasobe, S. Fukuzumi and P. V. Kamat, J. Phys. Chem. B, 2006, 110, 25477 CrossRef CAS; (c) T. Umeyama and H. Imahori, Energy Environ. Sci., 2008, 1, 120 RSC.
- T. Umeyama, N. Tezuka, M. Fujita, S. Hayashi, N. Kadota, Y. Matano and H. Imahori, Chem. Eur. J., 2008, 14, 4875 CrossRef.
- P. Brown, K. Takechi and P. V. Kamat, J. Phys. Chem. C, 2008, 112, 4776 CrossRef CAS.
- W. Lee, J. Lee, S. Lee, W. Yi, S. H. Han and B. W. Cho, Appl. Phys. Lett., 2008, 92, 153510 CrossRef.
- H. Z. Geng, K. K. Kim, K. P. So, Y. S. Lee, Y. Chang and Y. H. Lee, J. Am. Chem. Soc., 2007, 129, 7758 CrossRef CAS.
- L. Hu, G. Gruner, D. Li, R. B. Kaner and J. Cech, J. Appl. Phys., 2007, 101, 016102 CrossRef.
- N. Saran, K. Parikh, D. S. Suh, E. Munoz, H. Kolla and S. K. Manohar, J. Am. Chem. Soc., 2004, 126, 4462 CrossRef CAS.
- S. Berber, Y. K. Kwon and D. Tomanek, Phys. Rev. Lett., 2000, 84, 4613 CrossRef CAS.
- Z. Wu, Z. Chen, X. Du, J. M. Logan, J. Sippel, M. Nikolou, K. Kamaras, J. R. Reynolds, D. B. Tanner, A. F. Hebard and A. G. Rinzler, Science, 2004, 305, 1273 CrossRef CAS.
Footnote |
| † Part of the renewable energy theme issue. |
| This journal is © The Royal Society of Chemistry 2009 |