Metallomics: the concept and methodology
Sandra
Mounicou
,
Joanna
Szpunar
and
Ryszard
Lobinski
*
Laboratoire de Chimie Analytique Bio-inorganique et Environnement, CNRS UMR 5254, Hélioparc, 2, av. Pr. Angot, F-64053, France. E-mail: ryszard.lobinski@univ-pau.fr
First published on 7th January 2009
Abstract
The emerging field of metallomics refers to the entirety of research activities aimed at the understanding of the molecular mechanisms of metal-dependent life processes. This critical review discusses the concept of metallomics with a focus on analytical techniques and methods for the probing of interactions between metal ions and the organism’s genome and the derived -omes: proteome and metabolome. Particular attention is paid to the in vivo screening for the native metal–protein and metal–metabolite complexes by hyphenated techniques that combine a high-resolution separation technique (gel electrophoresis, chromatography or capillary electrophoresis) with sensitive elemental (inductively coupled plasma, ICP) or molecular (electrospray or MALDI) mass spectrometric detection. The contribution of bioinformatics to the prediction of metal-binding sequences in proteins and the role of molecular biology approaches for the detection of metal-dependent genes, proteins and metabolites are highlighted (115 references).
 Sandra Mounicou | Sandra Mounicou graduated from the University of Pau (France) from which she obtained a PhD in 2002. She was then a post-doctoral fellow at the University of Cincinnati, Ohio, before joining the National Research Council of France (CNRS) as a research scientist in 2005. Her research interests include the development of hyphenated techniques for the in vivo speciation analysis of metallocompounds, in particular metalloproteins, in biological systems. |
 Joanna Szpunar | Joanna Szpunar graduated from the Warsaw University of Technology, Warsaw (Poland) and then moved to the University of Warsaw where she obtained PhD (1992) and DSc (habilitation) (2000) degrees. She was a research fellow at the University of Antwerp (U.I.A.) (1993–1994), and a Fellow of the European Environmental Research Organization (EERO) at the University of Bordeaux (1995–1996). Since 1997 she has been working as a research engineer for CNRS and since 2007 is a titular professor of chemistry (Poland). Her research interests focus on analytical chemistry of metal–biomolecule interactions. |
 Ryszard Lobinski | Ryszard Lobinski is a research director at the CNRS and a professor of chemistry at the Warsaw University of Technology where he obtained PhD (1989) and DSc (habilitation) (1994) degrees. He has co-authored 3 books, over 200 research articles in international journals and given about 100 invited lectures. He was awarded a CNRS Silver Medal (2006) and is a Fellow of the Royal Society of Chemistry. His principal research interest is in the development of analytical approaches to species-specific (speciation) analysis for trace and ultratrace metals in environmental and nutrition chemistry and in life sciences. |
1. Introduction
The sequencing of the entire genome of the first free-living organism (Haemophilus influenzae) in 19951 and the completion of the human genome project in 20002,3 were the most land-marking eukaryote genome sequencing projects of over 500 under investigation during the last 15 years. The knowledge of the complete genetic blueprint of the increasing number of organisms has resulted in the emergence of different “-omics”, the disciplines aiming at the analysis of a particular class of components of a living organism in its ensemble.The genome of an organism contains information allowing the prediction of the entirety of the primary sequences of proteins that can be (but are not necessarily) expressed. This fact is the fundament of proteomics, the study of the complete set of proteins produced in a cell, tissue or organism, their localization, structure, stability and interaction.4,5 The exponential development of proteomics over the last decade became possible owing to the invention of soft ionization mass spectrometry (MS) techniques, using electrospray6 and matrix-assisted laser desorption (MALDI)7,8 ionization.
Every third protein is believed to require a metal cofactor, usually a transition metal, e.g.Cu, Fe, Zn or Mo.9 Metal ions may control up- and down-regulation of protein expression in cells, some proteins, e.g. metallothionein, being essential in homeostasis and detoxication processes.10 Metallochaperones protect and direct metal ions through the cytoplasm11,12 while extracellular albumin and transferrin are essential metallotransporters in human blood. The intracellular concentration of several metals, their distribution among the various cell compartments and their incorporation in metalloproteins are believed to be tightly controlled.13 The understanding of mechanisms by which a metal is sensed, stored or incorporated as a cofactor requires, in addition to the identification of metalloproteins, the characterization of the pool of non-proteinaceous molecules (products of enzymatic or biochemical reactions) interacting with metal ions or of metabolites of exogenous metallocompounds, e.g. metallodrugs. A systematic approach to the study of metal content, speciation, localization and use within organisms and ecosystems is becoming increasingly important.14
The canonical analytical approaches to proteomics and metabolomics usually ignore the existence of metal complexes with proteins and metabolites. The information on the metal–protein interactions is either lost during ionization (e.g.MALDI), on the level of sample preparation (because of denaturation) or simply not acquired because of the inadequate ionization efficiency, and, consequently, insufficient sensitivity. The characterization of the pool of metal-containing species in living organisms, the metallome, and of their interactions with the genome, transcriptome, proteome and metabolome requires dedicated analytical approaches to in vivo detection, localization, identification and quantification, in vitro functional analysis and in silico prediction using bioinformatics. The concepts of the metallome and metallomics and the discussion of the relevant analytical approaches are the topics of this critical review.
2. Metallome, metallomics and related terms
The term metallome was coined by Williams who referred to it as an element distribution, equilibrium concentrations of free metal ions or as a free element content in a cellular compartment, cell or organism.15 The latter would therefore be characterized not only by its genome or proteome but also by the metallome, their inorganic complement. The meaning of the term metallome was then proposed to be extended to the entirety of metal and metalloid species present in a cell or tissue type.16 The research field dealing with the study of metallomes and their correlations with genomes and proteomes was referred to as metallomics.17 Since then, metallomics has been the topic of a number of reviews,18–20 journal special issues21,22 and, last but not least, of a Royal Society of Chemistry journal dedicated to the field, Metallomics. The terms “metallome” and “metallomics” have been used in different contexts. In addition, a number of related definitions proliferated, e.g. ionomics,23 heteroatom-tagged proteomics24 or elementomics.25The term metallome used throughout this review refers to the entirety of metal and metalloid species present in a cell or tissue type, their identity, quantity and localization. It can be approached with different degrees of approximation, e.g. as a set of total element concentrations (multielement concentration blueprint, also referred to as ionome23) on the bulk level or at a specific location (e.g. a particular tissue or cellular compartment), or a set of metal complexes with a given class of ligands, e.g. metalloproteome (the entirety of metalloproteins) or metallometabolome (the entirety of metallometabolites). Metallome can be a static concept (the analysis will then have a specific endpoint per analogy to the genome, e.g. the determination of a cell multielement concentration blueprint, or the in silico prediction of a set of metalloproteins). It can also be a dynamic concept, per analogy to proteome or metabolome that responds in biologically important ways to environmental changes with an infinite number of possible variations. As there is no discrete proteome per se, even for simple single-cell organisms, there would be no discrete metallome either.
Metallomics is the study of a metallome, interactions and functional connections of metal ions and their species with genes, proteins, metabolites and other biomolecules within organisms and ecosystems. Metallomics has become a buzzword in recent years and abuse of the term is frequent. It is therefore important to specify that a metallomics study implies:
(a) A focus on metals or metalloids (e.g. As, Se, Sb) in a biological context. The extension of the term to biologically important non-metals, such as sulfur or phosphorous, is discouraged.
(b) A correlation of the element concentration blueprint or element speciation with the genome. This correlation may be statistical (an enrichment of an element coincides with the presence of a particular gene), structural (sequence of a metalloprotein is traceable to a gene) or functional (the presence of a bioligand is the result of a gene-encoded mechanism).
(c) A systematic, comprehensive or global approach. An identification of a metal species, however important, without specifying its significance and contribution to the system is not metallomics!
The different classes of metallospecies in a biological environment have been presented in Fig. 1. The most prominent of them include the metal complexes with proteins and with metabolites.
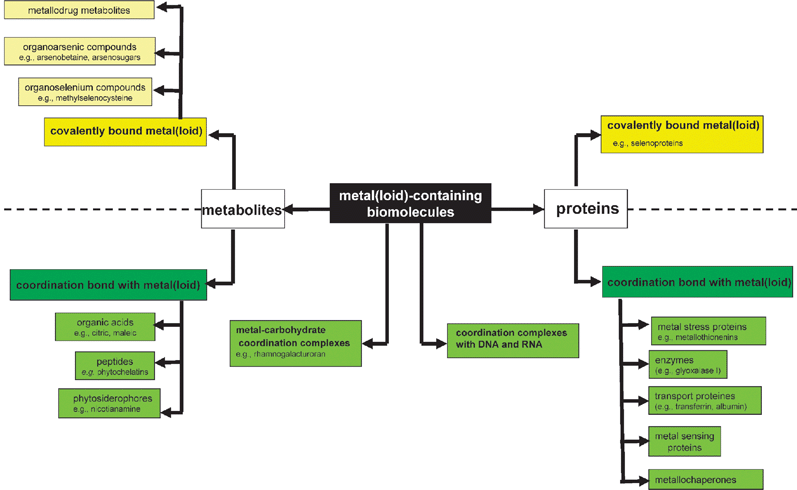 | ||
| Fig. 1 Metallome: the different classes of metal species in biological environment. | ||
A metalloprotein is defined as a protein whose function is conferred by a metal (the protein must bind a metal for its function). The function can be, e.g. a catalytic activity, implication in electron-transfer reaction of redox-active metal ions or stabilization of the protein ternary or quaternary structure. Metal is usually bound as a hydrated ion or a metal-containing cofactor (e.g.methylcobalamine). Metalloproteins should be distinguished from metal-binding proteins that simply complex a metal due to the existence of a thermodynamic equilibrium in the system; in such a case the metal has no particular function.
A metallometabolite is defined as a metal complex with an endogenous bioligand produced by a biochemical reaction or biosynthesized by an organism exposed to metal stress, or a biologically altered metallobiomolecule that had been deliberately introduced into an organism (e.g. as metallodrug).
3. Metallomics: areas of interest
Metallomics is a transdisciplinary research area with an impact on geochemistry, clinical biology and pharmacology, plant and animal physiology and nutrition. Its ultimate goal is to provide a global and systematic understanding of the metal uptake, trafficking, role and excretion in biological systems.Biological catalysis
A substantial number of enzymes require metals for their catalytic activity in fundamental biological processes, including photosynthesis, respiration and nitrogen fixation. The metals can occur as, usually hydrated, ions (e.g.Co2+, Cu2+, Fe2+, Fe3+ , Mg2+, Mn2+, Ni2+, Zn2+), be included in organic prosthetic groups, such as e.g.heme (Fe), molybdopterin or cobalamin (vitamin B12) or be incorporated covalently to form an enzyme active center, e.g.selenocysteine (SeCys) in glutathione peroxidase. The selection of a metal for use in enzymatic catalysis results from the combination of its physico-chemical properties, such as redox potential and coordination chemistry, and its accessibility in the environment for biological systems.26 The metal ions most widely involved in different mechanisms of substrate/factor activation in non-redox catalysis include Mg2+ and Zn2+ whose function consists in increasing electrophilicity, acidity or nucleophilicity of a reacting species or promoting heterolysis; Cu(I)/Cu(II), Fe(II)/Fe(III), Mn(II)/Mn(III) or Mo(IV)/Mo(VI) are prevailing redox couples in biological systems. The roles of metals in enzyme catalytic mechanisms were recently reviewed.26 The detection, quantitation, isolation and characterization of metalloenzymes is of fundamental importance in a number of disciplines, such as microbiology, plant and animal biochemistry and clinical chemistry.Biogeochemistry and environmental chemistry
The metals represent a link between the chemistry of the atmosphere and oceans from which life has evolved and the genomes and proteomes of living organisms. One aspect of evolution is due to the adaptation of the organisms to the changing environment in order to protect their reductive cytoplasmic chemistry and to obtain the required elements from the environment.27 The role of transition metals in microbial metabolism was discussed with a particular focus on the deduction of geochemical signatures and phylogenetic relationships of prokaryotes from whole genome sequences and on the use of this link to infer geochemical aspects of the biosphere through time.28 Model proteomes were reported to contain putative imprints of ancient shifts in trace metal geochemistry.29A systematic view of the transition metal metabolism requires an understanding of how organisms sense, adapt and use these metals within the biodiversity characteristic of each specific ecosystem.14 The environmental pollution with metals leads to the evolution of the protein expression signature and spurs the assignment of some proteins as environmental pollution biomarkers.19 The correlation of sentinel organism proteomes with the metal contamination is one of the areas of environmental proteomics.19
Plant biochemistry and physiology
The primary concern is the search for mechanisms responsible for the (i) mobilization of low solubility metals from soil, (ii) translocation within the plant, and (iii) sequestering metal ions in cytosol or in cellular compartments. Consequently, the identification and quantification of low molecular metal-containing metabolites is of interest in studies of:(i) Uptake and bioavailability of essential elements, mainly Fe and Zn. The related research aims at maximizing the transfer of these elements into grains and/or other edible plant tissues, in order to combat the micronutrient malnutrition that affects more than half of the world population, particularly in developing countries.30
(ii) Metal hyperaccumulation with a focus on phytoremediation and/or phytomining.31 Some plants known as hyperaccumulators accumulate and store metal ions in their aerial tissues at levels that are toxic to other species. Phytoremediation has appeared as a promising alternative in the removal of heavy metal excess from soil and water (based on the cultivation of aquatic plants) whereas phytoextraction can be used for the recovery of precious metals (phytomining).
(iii) Plant defence mechanisms against heavy metal stress (including volatilization, appearance of inorganic insoluble forms or induction of phytochelatins).32 The analytical focus is the investigation of the plant metabolome and the research for the implied genes and enzymes.
Clinical chemistry
Despite their vital role as regulatory cofactors, metals can also be highly toxic and be functionally associated with pathophysiology of several diseases. Most diseases associated with copper, iron and zinc result from mutations in the genes encoding for metal-transport proteins, which can be identified by proteomics approaches.33,34The increasing interest in the detection of biomarkers raises interest in the use of metalloprotein or metalloenzyme concentrations to identify the disease, track its progression, and help evaluate the impact of therapeutic agents. For example, matrix metalloproteinases, a family of zinc-dependent proteolytic enzymes that degrade extracellular matrix components, were reported as potential diagnostic indicators of cardiovascular disease.35 They are currently detected by quite tedious immunoessays to which metallomics analytical technologies may be an attractive alternative.
A number of pharmaceuticals contain a metal in their structure and are referred to as metallodrugs.36 They have anticancer function (e.g.cisplatin or Ru compounds), antiarthritis (Au), gastric (Bi) and others. Metallomics will study the metabolism of these drugs, transport and interactions with biologically relevant molecules.
Nutrition and essential element supplementation
The essential role of many trace elements, their implication in prevention of a number of diseases and the low levels of some of these elements in the diet in many countries are at the origin of the increasing interest in the supplementation of food and feed with mineral elements. The most popular element of concern has been selenium, supplied in the form of yeast grown in the presence of selenite,37 but supplements for other metals, e.g.Zn, Fe, Mn and Cr, proliferate on the market. The relevant metallomics studies focus on two levels:(i) The characterization of the chemical forms and their bioavailability in the supplements. The knowledge of virtually all the forms present at levels exceeding 0.1% is required to comply with regulations of governmental agencies issuing sales authorizations. The elemental speciation pattern is also a precious tracer of the origin of the product and its control allows a better optimization of the biotechnological production process.
(ii) The transport and fate in the target organisms, such as animals (meat, milk and eggs from Se-supplemented animals become functional food themselves) and humans.38
4. Experimental approaches to metallomics
A number of analytical approaches have been proposed for metallomics. They can be divided into three basic categories, in vivo, in vitro and in silico as illustrated in Fig. 2, and include:(a) Methods for the analysis for metal-containing biomolecules present in a biological sample.
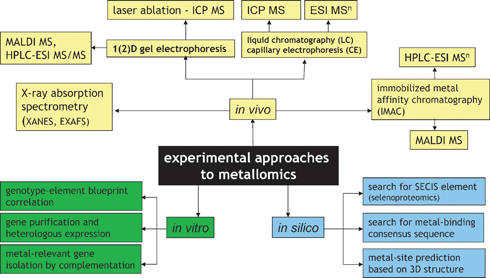 | ||
| Fig. 2 Experimental approaches to metallomics. | ||
In this approach, a metallospecies is made to produce a specific signal allowing its detection, identification and quantification. Because any removal of the analyte from its original environment bears the risk of altering its identity, techniques that can be directly applied to the sample should be used. The most attractive is X-ray absorption spectroscopy (XAS), which can provide some useful data on the metal-coordination environment and oxidation state but the high level of metal concentration and sample purity required limit its applications to very few free-leaving organisms, e.g. hyperaccumulating plants.
The trace level concentrations of metalloproteins and metallometabolites present can be addressed by hyphenated techniques that combine a high-resolution separation by chromatography or electrophoresis with sensitive detection by elemental or molecular MS. The role of the separation component is to provide sufficient resolution to avoid the co-elution of another species of the same element (when ICP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) MS detection is used) or that of any easily ionizable concomitant species capable of suppressing the analyte’s ionization (when molecular MS is used).
MS detection is used) or that of any easily ionizable concomitant species capable of suppressing the analyte’s ionization (when molecular MS is used).
The use of hyphenated techniques requires the metal species to be kinetically inert, i.e. not to engage in reactions that result in replacement of one or more ligands in its coordination sphere by others on the timescale of the analytical procedure. The thermodynamic stability (the feature of the species to exist under equilibrium conditions) should also be taken into account when designing extraction and chromatographic mobile phase conditions.
The stability and recovery concerns during chromatography should be verified, ideally by collecting the metal-containing compound fraction and repeating the analysis using the same experimental conditions. Whereas precautions during chromatography are never too many, the primary source of species modification is the extraction step itself. It must be admitted that a number of metallocompounds cannot be characterized in vivo at all with the currently existing methods, either because of insufficient concentration, purity or stability.
A class of in vivo analytical methods does not target intact metal complexes but simply biomolecules showing an affinity to bind metals. Such molecules are typically isolated by immobilized metal affinity chromatography (IMAC). The analysis is qualitative and concerns the ligand only. The existence of the metal–ligand bond in vivo is assumed on the basis of the characteristics of the separation step.
(b) Methods based on the in vitro modification of the organism’s DNA (or isolation of particular genes) and determination of the impact of this operation on the metallome.
The most common approaches are: (i) building up a large collection of mutants (differing by deletion of a particular gene) of a model organism (e.g. Arabidopsis thaliana or S. cerevisiae) and screening them for the abnormality in element concentration blueprint in order to detect metal-regulated genes, (ii) purification of a large number of open reading frames (ORFs) from a given organism, heterologous expression of proteins and screening for those that contain metals, and (iii) screening of gene libraries (banks) on metal-toxic media in order to detect genes implied in metal resistance.
(c) In silico approaches.
They consist in searching for metal-binding motives (which are expected to be preserved regardless of the organism studied) in putatively expressed proteins based on genome sequences present in data banks or for other patterns (e.g. SECIS) relevant to the function of heteroelements.
5. Proteomics and analysis for metalloproteins and metal-binding proteins
Proteomics analysis aims at the high-throughput identification and quantification of the proteins present in the sample. The typical proteomics strategies profit from the knowledge of the studied organism genome, and consequently the set of the putatively present proteins, and include:(i) Bottom-up proteomics based on the 2D gel electrophoresis followed by the tryptic digestion of spots and MS analysis.39 The identification of a few peptides is generally sufficient to identify a protein present.
(ii) Shotgun proteomics.40 The sample is digested with trypsin and the resulting peptides are separated by 2D nanoHPLC and identified with ES MS/MS. The comparison of the data set with a set or proteins potentially synthesized by a given organisms allows the identification of the proteins present.
(iii) Top-down proteomics.41,42 The proteins are separated by LC and fragmented by infrared multiphoton dissociation (IRMPD) or electron capture dissociation (ECD). The amino acid composition of the fragments is identified by high-accuracy MS. The identification is based on correlations between the amino acid compositions of the formed ions and the set of protein possibly present.
The specificity of metalloproteomics studies demands the need for the description of the metal-binding sites, metal stoichiometry and metal-dependent structure or conformation changes. This is not compatible with protein denaturation, which usually results in the loss of the non-covalently bound metal cofactor. A valid metalloproteomics protocol requires: (i) the preservation (or reconstitution) of the link between the protein and the metal, (ii) development of a filter allowing the specific detection of metalloproteins in the sample and (iii) purification and/or preconcentration of metal-binding proteins of concern. These requirements result in two types of approaches for the analysis of proteins containing a non-covalent metal cofactor:
(i) Extraction of the entirety of the sample proteome (without caring for the metal–protein bonds) followed by the isolation of the set of proteins with affinity to a particular metal and their identification by one of the canonical proteomics approaches. This approach detects proteins with metal-binding affinity in the sample but does not specify whether the metal complexes were actually present and with which proteins,
(ii) Extraction of intact metal–protein complexes followed by their separation and identification. The emphasis is made here on the preservation of the metal–protein complex. The link can be broken only when the degree of purity is achieved that allows the certitude of the existence of the metal–protein bond before denaturation.
5.1 Identification of proteins with a covalently bound metal
Proteins containing a covalently bound heteroelement (e.g.Se) are amenable to any of the three canonical proteomics approaches discussed above. The metal-bearing protein or peptide can be recognized by its isotopic pattern, but in practice, its signal is suppressed by the co-elution of the more abundant, easier ionizable concomitant species. The particularity of the metallomics approach consists of the parallel specific ultrasensitive and quantitative detection of only those proteins/peptides that contain a heteroatom, regardless of their abundance and of the presence of the accompanying matrix. This detection is carried out by ICP MS and can be incorporated at different stages of the proteomics canonical procedures, as illustrated in Fig. 3. Consequently, parameters such as the separation recovery and the digestion efficiency can be quantitatively monitored and the subsequent molecular MS targets the metallomolecules only.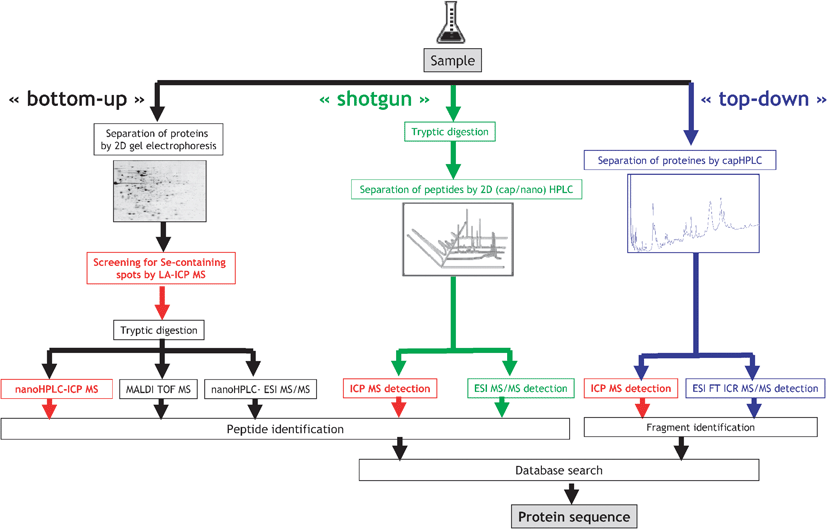 | ||
| Fig. 3 Element-specific detection in proteomic canonical protocols. | ||
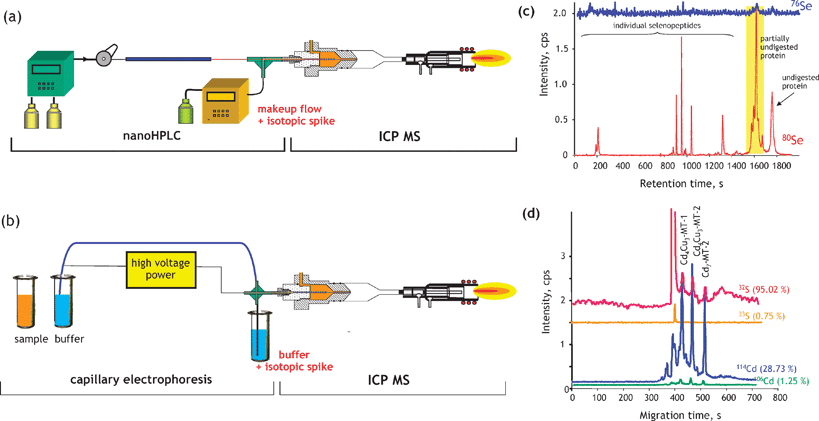
The identification of heteroatom-labelled proteins by the bottom-up or shotgun strategy is carried out using an experimental setup (Fig. 4a) that allows the acquisition, in addition to the conventional total ion current chromatogram by nanoHPLC-electrospray MS/MS (Fig. 4b), of an element specific chromatogram by nanoHPLC-ICP MS (Fig. 4c).43,44 The approach, discussed here for the analysis of Se-containing proteins in Brazil nuts,44 allows:
(i) The determination of the retention time at which Se-peptides elute. Consequently, the number of mass spectra in which the molecular mass peak (isotope characteristic envelope) should be found is reduced from a few thousands to a few, which is important because of the absence of machine algorithms allowing the search of heteroatom-containing species. Note that the failure to find a signal in the spectrum where it should be found would mean the insufficiency of preconcentration/purification. In the presented example, Se compounds were detected in 6 out of 8 mass spectra expected to contain them (Fig. 4d). The extracted ion chromatogram for these ions corresponds to that with the Se-specific ICP MS detection (Fig. 4e).
(ii) The determination of the mass balance of the Se-species owing to the ICP MS signal being quantitative. This allows the determination of the tryptic digestion efficiency (amount of non-digested and partly digested protein, cf. section 9), the determination of the column recovery and the monitoring of the purification efficiency.
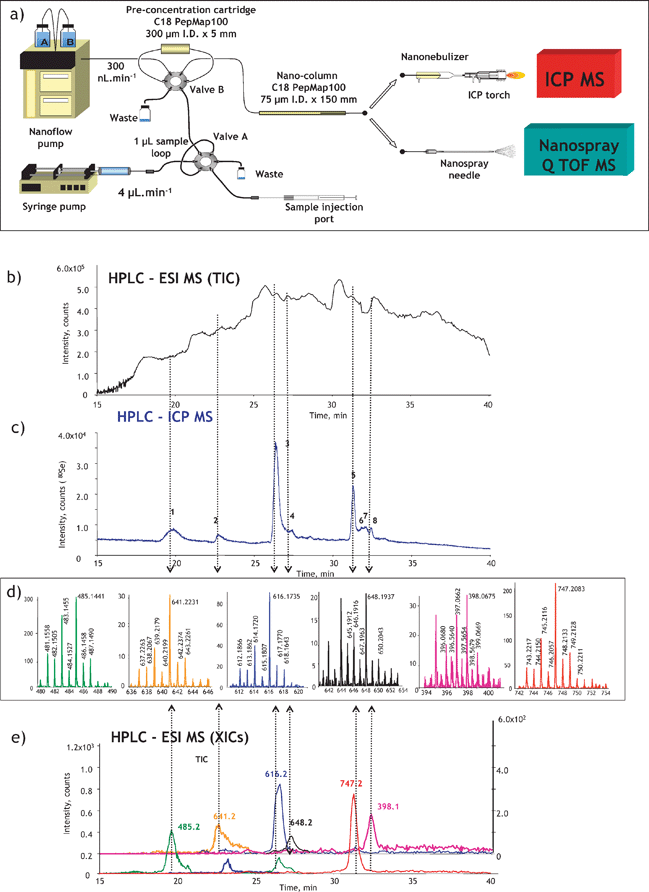 | ||
| Fig. 4 ICP-MS-assisted shotgun proteomics approach. (a) Experimental setup; (b) a total ion current HPLC-ESI MS chromatogram; (c) HPLC-ICP MS chromatogram (Se-specific); (d) selenium isotopic patterns found in ESI mass spectra corresponding to the selenium ICP MS peaks; (e) extracted ion ESI chromatograms for Se-containing peptides. The example concerns the analysis for Se-containing proteins in Brazil nuts.44 | ||
5.2 Identification of proteins with metal-binding affinity
The principal technique used to capture proteins with metal-binding ability is immobilized metal affinity chromatography (IMAC). Possibilities of the use of immunoaffinity chromatography using a metal catalytic domain antibody to fish out target metalloproteins from the tissue (i.e. human serum and plasma) were evoked but practical realization of this idea was not reported.35 Another elegant trend can be the use of metal-coated IMAC chips in SELDI.45IMAC resins use chelating agents (e.g. tridentate iminodiacetic acid) immobilized on a sorbent. Prior to use, metal ions are immobilized on the IMAC resin and are supposed to react with specific protein ligands in the sample. An IMAC protocol includes the saturation of an IMAC support (column/chip/chip array) with the metal of interest, washing off the excess metal, introduction of a metal-depleted sample (metal depletion results in unoccupied metal-binding sites of proteins, which facilitates binding to the IMAC metal) and the removal of the non-bound sample components followed by the elution of the retained proteins (with the metal-binding affinity) with a competing complexing agent. The proteins with the metal affinity are recovered as apo(demetallated forms) and can be analyzed by any of the classical proteomics approaches.
Alternatively, the retained proteins can be digested on-column. The metal-coordination center was expected to be preserved by the retention of the metal–peptide complex after the digestion of the protein,46,47 which seems to be a convenient way for the identification of the metal-binding motives.
The IMAC approach has enjoyed a growing popularity, with recent applications referring to the analysis of the zinc46,47 and copper45–47 proteomes in human hepatoma cell lines, uranyl proteome in human serum,48bismuth proteome in Helicobacter pyloricell extracts,49 and Ni-proteome in human keratinocytes34,50 and Arabidopsis thaliana roots.51
IMAC does provide information on the presence of proteins with metal-binding sites but not on the metal–protein complexes present. Also, metalloproteins with a high metal affinity at functional sites, e.g.superoxide dismutase (SOD), are likely to pass through the IMAC column due to occupation of binding sites by physiological metals.47 The IMAC approach is not quantitative and minor proteins are likely to be masked by major ones.
5.3 Screening for metal–protein complexes by gel electrophoresis with laser ablation ICP MS detection
Polyacrylamide gel electrophoresis (PAGE), employed in either the monodimensional or 2D mode is considered the most adequate technique for the separation of proteins. The principal difficulty in its use for metalloproteomics is the need for the preservation of the metal–protein bond. Many metal-complexes with proteins are labile and can be destroyed by exchange with the metal impurities of the gel during separation and staining. Amongst the recommended precautions, the most important are: (i) the use of non-denaturating separation protocols, (ii) avoiding the presence of metal impurities in gels, and (iii) avoiding staining or the use of ultrapure staining reagents (e.g. in BlueNative electrophoresis52).Metal-specific detection in the gels has enjoyed considerable interest for a long time, the principal techniques including autoradiography with its inherent use of radioactive isotopes (e.g.75Se), and synchrotron radiation XRF and PIXE with the need for hardly available facilities. Laser-ablation (LA)-ICP-MS detection, pioneered by Nielsen et al.,53 offers a competitive alternative for the in situ probing of the protein spots for the presence of metals and metalloids. The ablated analytes are swept into the ICP by a continuous stream of argon, and the ions are analyzed by MS. As a result, an electropherogram is obtained in which the quantity of a given element is a function of its position in the gel. Detection by LA-ICP-MS is a potentially fast and fairly robust technology, because no further reaction or derivatization step is involved, and the signal is, theoretically, directly proportional to the quantity of the analyte element in the gel.54LA-ICP-MS detection can be carried out in three principal modes presented in Fig. 5. They are:
(i) Lane scan (Fig. 5a). As the whole gel is scanned, the metalloproteins do not need to be visualized by staining thus minimizing the risk of metal loss.55
(ii) Spot-to-spot hopping (Fig. 5b). A disadvantage of this technique is that protein spots must be visible in order to be ablated.56
(iii) Imaging mode (Fig. 5c).57 The high stability and precision of LA-ICP-MS make it possible to scan a gel in raster mode and thus to acquire element images. The analysis in the imaging mode is time consuming. Scanning a gel of 5 cm2 with a 100-μm resolution takes about 15 h but the technique allows the identification of areas with increased metal concentrations regardless of the presence of intense protein spots. As the whole gel is scanned, the metalloproteins do not need to be visualized by staining.
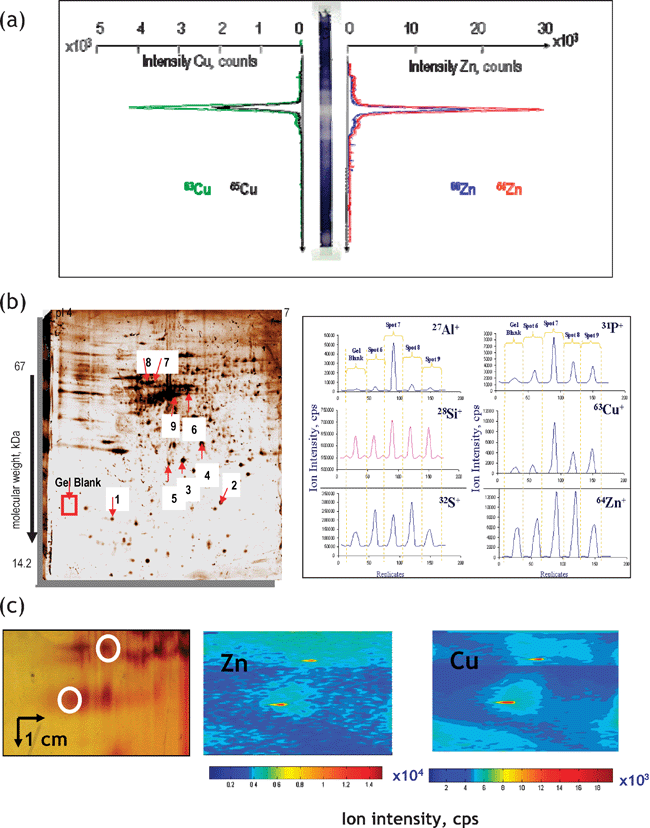 | ||
| Fig. 5 Principal modes of data acquisition in gel electrophoresis laser ablation – ICP MS. (a) Lane scan (the example concerns detection of Zn and Cu in superoxide dismutase isoforms. Reprinted with permission from ref. 55; (b) spot-to-spot hopping. The example concerns the analysis of metal-containing proteins in human brain. Reprinted with permission from ref. 56 (c) imaging mode. The example concerns the analysis of metalloproteins in rat kidney extract.57 | ||
Proteins containing covalently incorporated metals (e.g. selenoproteins) can be recovered from the gel and analyzed using the approach described in section 5.1.58 The recovery of an intact metal–protein complex is much more difficult. Unless a mass spectrum of the metal–protein complex is provided there is always a risk that the protein identified by the spot digestion and peptide mapping is different from that binding the metal in the same spot. Auxiliary data such as the presence of metallated peptides in mass spectra, in-gel EXAFS showing the metal–protein bond59 or the desorption of the metalloprotein from the gel (or the blot) are required.
5.4 Chromatographic analysis for metal–protein complexes
The difficulties with controlling the stability of metal–protein complexes and with their recovery from the gels for further analysis in gel electrophoresis spur interest in chromatographic techniques for metallomics. The most favored tool for a rapid semi-quantitative screening for the presence of metal–biomolecule complexes in biological samples has been the coupling of size-exclusion LC and ICP MS (for reviews of applications see refs. 18 and 60). The elution is monitored in the multielement (multiisotopic) mode, and as the column can be calibrated in terms of molecular mass, a chromatogram (Fig. 6a) allows the fractionation of the metallobiomolecules as a function of size prior to detection. Fairly concentrated samples (e.g. 3–5 times diluted cytosol) can be analyzed and conditions for non-denaturating separations can be readily optimized.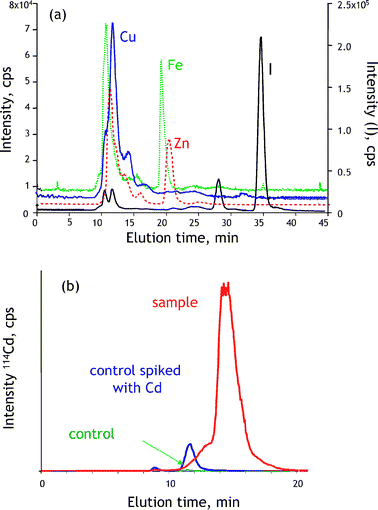 | ||
| Fig. 6 Liquid chromatography with ICP MS detection in metallomics. (a) Multielement screening for metal-containing species in human milk. (b) Detection of bio-induction of Cd-binding proteins in rat tissue. | ||
In practical terms, a typical metallomics experiment (Fig. 6b) by SEC-ICP MS involves three analyses: one of a control sample, one of a control sample spiked with excess metal (to evaluate the metal-binding capacity and the presence of endogenous metal-binding ligands) and one of a sample that had been exposed to metal stress. A difference between the chromatogram of the latter and the chromatograms of the control sample and of the metal-spiked one allows the detection of the functional metal stress-induced protein complex.
The resolution of SEC is low and the chromatographic purity of peaks is usually poor. However tempting it may be, the matching of the elution volume with that of a standard cannot be considered as a definitive proof of the species identity. Also, the control of the adsorbed metal ions on the stationary phase is important as they can exchange with the metal ions already complexed or can be scavenged by ligands from the sample leading to ghost peaks.
The detected metallocomplexes can be identified in terms of molecular mass by electrospray MS. The ionization efficiency is critically dependent on the analyte chromatographic purity and can be problematic. Even if a single species of a given element is present (as detected by ICP MS), hundreds of matrix compounds, transparent to the ICP MS detection may co-elute. Therefore, a purification step using an orthogonal separation mechanism is required prior to electrospray MS.61ICP MS offers the possibility of monitoring the purification of the metalloprotein while accounting for its integrity (monitoring of the metal or, in some cases, the metal-to-sulfur ratio).
The principal advantages of chromatography over gel electrophoresis include the facility of the monitoring of the recovery and of the stability of a metal–protein complex during separation and the versatility allowing the analysis of other types of metallomolecules. The metal-containing fraction can be readily collected for further analysis and the stability of the metallocomplexes present can be checked by repeated chromatography under the same conditions. Column techniques (chromatography or capillary electrophoresis) with the parallel ICP MS and ESI MS detection seem to offer the most adequate tool for the in vivo identification of metal–biomolecule complexes at the pico- and nanomolar levels although the number of successful identifications to date has been limited and concerned relatively small proteins, such as metallothioneins.
6. Metallometabolomics
A successful approach to metallometabolomics should provide evidence of the identities of all the species of a given element present, or at least identify some and account clearly for the number of non-identified ones (in other words, produce a quantitative speciation blueprint). In contrast to metalloproteomics, there exists no direct metabolite–gene relationship, so any identification of metabolites has to be carried out de novo and requires advanced high-resolution molecular mass spectrometric techniques. Metabolites are often hydrophilic as the metabolic process generally results in the addition of polar groups to enhance elimination from the cellular tissue. Unlike for peptide analysis, desalting (and separation) protocols for small hydrophilic molecules are not well developed, not to speak of relevant protocols for metal–metabolite complexes.In some cases, the metallometabolome can be probed directly by XAS (X-ray absorption spectrometry), as e.g. in the case of hyperaccumulating plants when one or two dominant metabolites complexing a metal are present.62 Otherwise, extraction of species of interest is required; it is followed by a chromatographic (or electrophoretic) separation, detection of metallospecies by high-accuracy MS and their subsequent identification by multistage MS.
6.1 Direct analysis of metallospecies by XAS
XAS, also called X-ray absorption fine structure (XAFS), is a generic term for techniques based on the analysis of a spectrum obtained by absorption by an atom of X-ray photons, produced by a synchrotron source and the measurement of the resulting fluorescence intensity. The X-ray absorption spectrum is presented as a function of the X-ray energy and can be divided into two energy regions, the edge region and the beyond the edge region (Fig. 7a). Analysis in the edge region is usually referred to as XANES (X-ray absorption near edge spectroscopy) and in the beyond the edge region as EXAFS (extended X-ray absorption fine structure region).63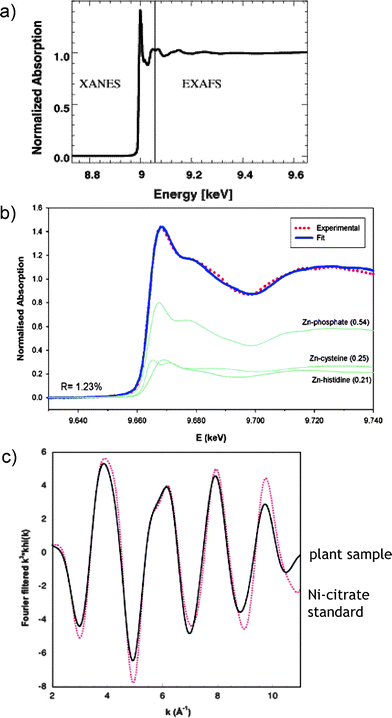 | ||
| Fig. 7 Analysis of metal-binding in plants by X-ray absorption spectrometry (XAS). (a) A typical XAS spectrum showing the XANES and EXAFS regions. Reprinted with permission from ref. 62; (b) XANES data fit showing the distribution of Zn into phosphate, cysteine and histidine complexes in leaves. Reprinted with permission from ref. 64; (c) EXAFS data fit confirming the identity of the Ni-citrate complex in a hyperaccumulating plant. Reprinted with permission from ref. 65. | ||
XANES (the energy of the edge and the assignment of peaks near and on the edge) gives information about oxidation number, covalence (the number of pairs of electrons that a given atom shares with its neighbors), molecular symmetry of the site and, hence, coordination number. The EXAFS provides structural information about the atomic neighborhood of a metal (metalloids) being probed, such as the coordination number (number of ligands), the identity of the ligand atoms, and the distance between the metal and each ligand.
XAS is performed directly on the sample, even non-crystalline, without preliminary extraction or preparation. It is a quantitative technique but it is not particularly sensitive (a minimum analyte concentration at several tens of ppm level is required). In the case of a mixture of different species of the probed element present, the interpretation of the spectra becomes rapidly difficult, so the technique is ideal when a single (or maximum two) dominant species is present. Identification is carried out by the comparison with the standard of the oxidation state or coordination center.
In metallometabolomics, XAS is invaluable for probing the kinetically labile complexes that can readily dissociate or exchange ligands during sample handling and for the differentiation of the oxidation state, which in some cases [e.g.As(III)/As(V)] is readily modified during any extraction step. In an example shown in Fig. 7b, XANES allowed the determination of the quantitative distribution of Zn among the complexes with phosphate, cysteine and histidine in plants.64Fig. 7c shows the match of EXAFS spectra allowing the identification of citrate as the major Ni-binding ligand in Leptoplax leaves.65
XAS, which is becoming popular owing to a rapid democratization of the access to synchrotrons, gives the comfort of avoiding sample preparation but its areas of application are limited. It can only confirm the presence of expected species in a sample but cannot find new ones. Comparative studies between the XAS and hyphenated techniques proved the conservation of the original species in carefully optimized sample preparation protocols.66
6.2 Mass spectrometric pathways to identification of metallometabolites
Molecular MS offers unique advantages for the identification of metabolites at the sub-nanomolar levels. Electrospray has been the preferred method of ionization although the use of atmospheric pressure chemical ionization (APCI) and MALDI (except the region of interference with matrix ions and non-covalent species) were also reported. Metabolite structures can be elucidated on the basis of the empirical formulas of the parent compound and fragment ions (data provided by high-resolution and high-accuracy MS) and of the lineage of fragment ions observed in tandem MS and, especially, in multistage MS (MSn).67,68(i) The species itself (some metallocomplexes are inherently difficult to ionize and little can be done about it!).
(ii) Ionization conditions (for a given species a trade-off between the conditions for optimum ionization and the species thermodynamic stability is often necessary; metal-complexes may not be stable in the acidic pH preferred by electrospray).
(iii) The buffer concentration (low concentrations of volatile buffers, e.g.ammonium formate, must be used).
(iv) The presence of concomitant easily ionizable species (an intense ion from an interferent entering the mass spectrometer at the same time as the analyte) will suppress either the ionization or the mass analysis (the intrascan dynamic range of the mass analyser is vitally important here).
For the above reasons electrospray MS analysis is never carried out directly on a sample extract.
The probability of successful identification is increased by sample introduction by HPLC as demonstrated by the identification of Ni species including malate, histidine, citrate and nicotianamine complexes in A. thaliana xylem sap,69,70 complexes of Cu and Zn with 2′-deoxymugineic in wheat shoot and roots press sap70 or phytochelatin–cadmium complexes from plant tissue cultures.71 However, straightforward HPLC-ESI Q-TOF MS/MS often gives incomplete results, failing to detect a number of species present. For example, when applied recently to a study of the metabolome of selenium-rich yeast, it allowed the detection of only 4 Se species out of more than 20 species present.72
Therefore, a comprehensive approach to metallometabolomics requires, at the present state of MS technology, a purification step.73 The elution of metallometabolites is ideally monitored by ICP MS of which the sensitivity is independent of the chemical environment of the metal(loid) in the molecule, chromatographic buffer and the co-eluting matrix or concomitant compounds. In addition, ICP MS allows the quantitative monitoring of recovery of the analyte, an essential parameter that escapes ESI MS measurement. Note that ICP MS cannot monitor the quality of separation per se but it is assumed that analyte purity increases with each separation step when clean volatile buffers are used.
The high polarity and low hydrophobicity of metallometabolites make aqueous normal phase chromatography, also called hydrophilic interaction chromatography (HILIC)74, an ideal sample introduction technique for metallometabolomics by electrospray MS. The columns employed usually contain polar/hydrophilic groups in the stationary phase (amido, cyano, amino groups).74 The mobile phases commonly used (acetonitrile, methanol, low concentrated and volatile aqueous buffers) are compatible with the use of an electrospray source. Retention times increase with the hydrophilicity of the solutes and most of the metal complexes can thus be retained in contrast to reversed-phase LC. The metal-related applications of HILIC include the analysis of the yeast selenometabolome68 and of metal complexes with small organic molecules such as divalent-phytosiderophores [Zn, Cu, Ni but apparently not Fe(II)]69,70 and organic acids (Ni malate, citrate, histidine, EDTA and nicotianamine).73
| Formula | Calculated mass | Error (mDa) | Error (ppm) | RDB |
|---|---|---|---|---|
| Error <1 ppm | ||||
| C16H35O13P2Se | 577.071265 | −0.03544 | −0.06141 | 1.5 |
| C9H25N10O14Se | 577.071143 | 0.08704 | 0.15083 | 3.5 |
| C17H29N4O11SSe | 577.071327 | −0.09695 | −0.16801 | 6.5 |
| C6H34N4O19PSe | 577.071461 | −0.23087 | −0.40007 | −6.5 |
| C14H31N10O2P2S2Se | 577.070788 | 0.44208 | 0.76607 | 6.5 |
| C9H33N6O13S2Se | 577.070676 | 0.55379 | 0.95966 | −2.5 |
| Error <2 ppm | ||||
| C8H39N2O15P2SSe | 577.070615 | 0.61530 | 1.06625 | −7.5 |
| C9H35N6O11P2SSe | 577.071952 | −0.72201 | −1.25116 | −2.5 |
| C19H34N2O7PS2Se | 577.070481 | 0.74922 | 1.29832 | 5.5 |
| C10H29N10O9S2Se | 577.072014 | −0.78352 | −1.35775 | 2.5 |
| C17H39O8P2S2Se | 577.072136 | −0.90600 | −1.56999 | 0.5 |
| C11H30N8O10PSSe | 577.070297 | 0.93322 | 1.61716 | 2.5 |
| C7H38N4O14PS2Se | 577.072331 | −1.10143 | −1.90866 | −7.5 |
Note that mass accuracy is the function of the characteristics of the mass analyser itself but also of the signal-to-noise ratio, which can be critical during the analysis of real-world sample extracts. Instrument manufactures claim values below 2 ppm for the latest TOF mass analysers using mass-lock calibration but they are obtained for pure analytes in ideal conditions. In practice, for the analyses of biological extracts, the mass accuracies reported were not lower than 10–20 ppm. Hence, the empiric formula determination requires a mass analyser using fast Fourier transformation to obtain mass spectra such as FT ion cyclotron resonance or an electrostatic ion trap (Orbitrap). Their advantage is a dynamic range of more than 5000, which is at least an order of magnitude higher than typical values reported for time-of-flight instruments.
A systematic de novo identification of metallometabolites can be achieved by high-resolution multistage MS (MSn), which is possible in an ion cyclotron resonance or an electrostatic ion trap (Orbitrap). An example of the measurement is shown in Fig. 8.68 The empirical formula of the parent ion is determined by the exact mass measurement and the parent ion is fragmented. Owing to the sub-ppm mass accuracy the empirical formula of the product ions can also be determined. The product ions can be further broken down to fragments that are sufficiently small to be unambiguously identified by their empirical formula only. The data interpretation is based on the analysis of the fragment–parent ion lineage up to the molecular ion. An advantage of FT MS over TOF MS is the preservation of the high mass accuracy in mass spectra up to MS4 while the large fragmentation window allows following the lineage over the whole m/z range of the isotopic pattern of the metallomolecule.
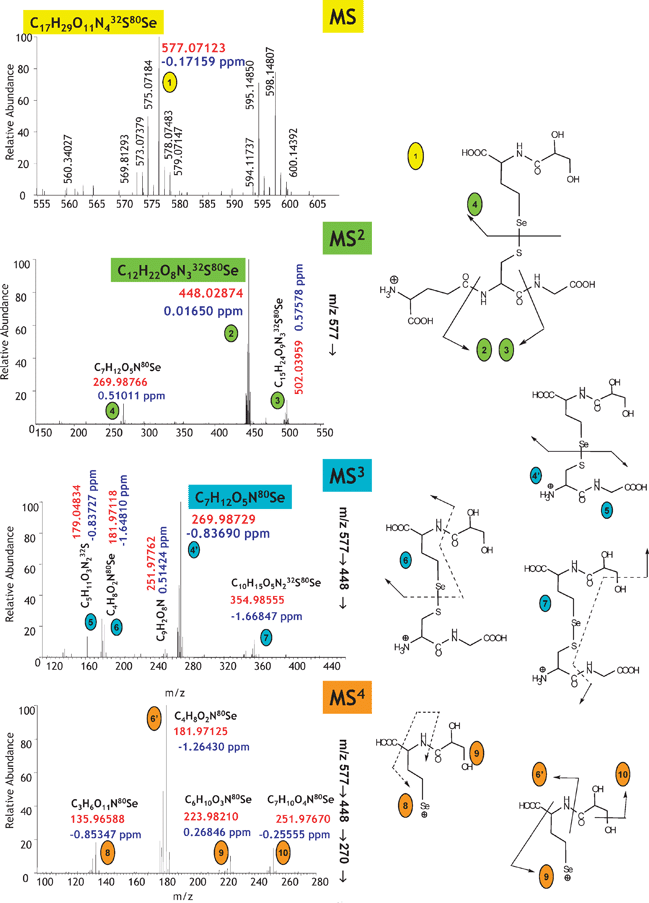
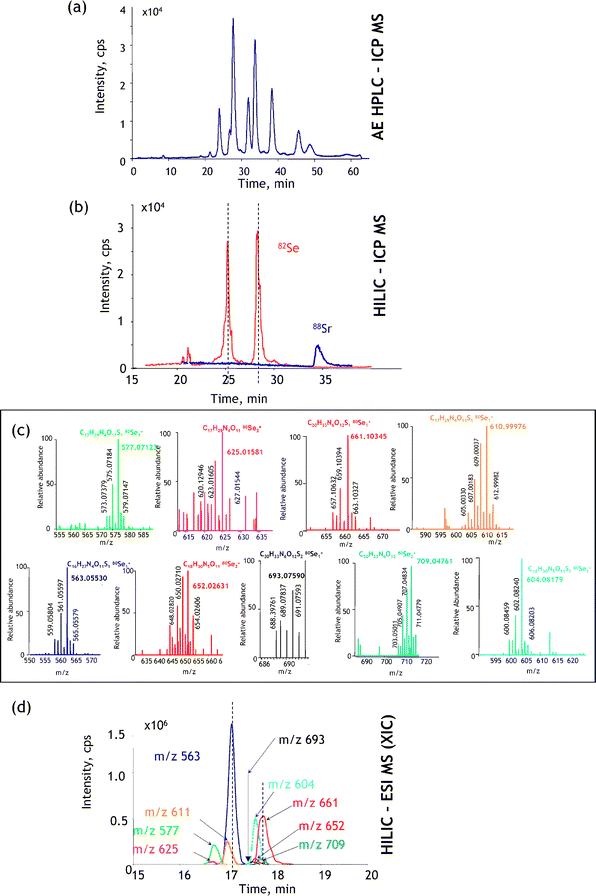 | ||
| Fig. 9 A generic approach to metallometabolomics. The example concerns the identification of a Se-metabolite in Se-rich yeast.68 (a) Verification of the number of heteroatom-containing species present by maximum resolution (in this case anion-exchange) LC-ICP MS; (b) verification of the quantitative recovery and retention times by HILIC-ICP MS (88Sr is monitored as a marker of the retention time of the elution of salts); (c) identification of the Se isotopic patterns at the relevant retention time by ESI MS; (d) reconstruction of the ESI FT MS extracted ion chromatograms. Reprinted with permission from ref. 68. | ||
The first step consists of determining the number of metallometabolites present and proving that the sum of their concentrations accounts for the total concentration of the probed element. ICP MS detection in HPLC or capillary electrophoresis offers an adequate tool to optimize the separation for the maximum number of resolved compounds while monitoring continuously the species recovery and, to a large degree, their stability on the column. In contrast to ESI MS, the choice of the separation mechanism and conditions is not compromised by the signal suppression or insufficient sensitivity, so a blueprint of all the metallospecies present can be obtained (their thermodynamic stability and kinetic inertness permitting). In the example shown in Fig. 9a anion-exchange HPLC could be optimized to show the presence of 9 selenium species (accounting for 98% of the total Se present). Note that ESI MS would not produce a signal under these conditions because of the use of a relatively concentrated buffer required to obtain the high separation efficiency.
The second step consists of finding a separation technique that still allows the quantitative recovery and the maximum resolution of the analyte compounds, but, imperatively, in optimum conditions for the electrospray ionization. This is best achieved by HILIC; ICP MS detection is necessary to verify the quantitative species recovery. A chromatogram for the case studied is shown in Fig. 9b. The resolution is worse than in the case of the anion-exchange separation but the composition of the mobile phase enables sensitive ESI MS detection.
The third step (Fig. 9c) includes the acquisition of a HILIC-FTMS chromatogram and data mining in order to find the 9 selenium species known to be present. The information acquired by anion-exchange HPLC-ICP MS tells us how many species should be detected before data analysis (which is a complex manual process) can be considered to be complete. The HILIC-ICP MS chromatogram facilitates the data mining as it indicates the retention time region in which selenium compounds should be searched for. The insufficient number of Se compounds found suggests the inadequate fractionation/purification during the sample preparation step.
The experiment is completed by the acquisition of the MSn data (as shown in Fig. 8) for each of the compounds detected and reconstitution of the fragmentation pathways leading to the identification of the fragment and parent ions.
7. Analysis for metal–gene relationships
7.1 Effect of the organism genotype on the elemental blueprint
In this approach, schematically shown in Fig. 10, the effect of the presence or absence of a particular gene on the increase or decrease of the metal concentration in an organism tissue(s) is studied.23,77,78 A collection of 4000–6000 mutants of an organism is created, brought to a given growth stage and tissue(s) of each of them is (are) analyzed for a number of elements. A comparison with the corresponding concentrations in a control sample detects mutants enriched or impoverished in a particular element that can be correlated with a particular gene. The approach allows an insight into gene networks involved in mineral ion accumulation in plants. It requires a collection of mutants, for the moment available only for a limited number of model organisms, and high-throughput reliable multielement analysis, which can be achieved by ICP MS (in some cases assisted by ICP AES).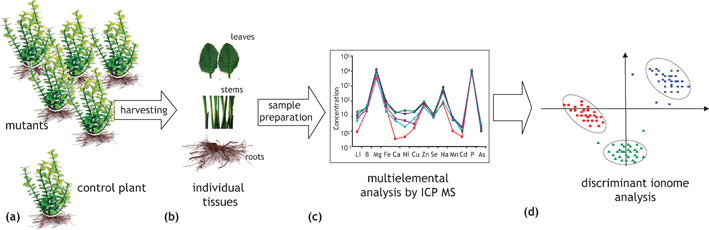 | ||
| Fig. 10 A schematic representation of a high-throughput ionomics experiment (search for the metal distribution—gene correlations). (a) Mutants and wild-type plants are grown to the same size under standardized conditions and are harvested at the same time. (b) Equivalent amounts and parts of plant tissues are sampled. (c) Multielement analysis of digested samples is carried out using ICP-MS and/or ICP -AES to identify changes in ionomes. (d) Data are collated and analyzed in order to correlate changes in ionome with a genome modification. According to ref. 23 and 76. | ||
The biological significance of connections between a genome of an organism (Arabidopsis thaliana23 and Saccharomyces cerevisiae78) and its elemental profile could be demonstrated. The range of trace elements investigated included well-known enzymatic cofactors: Mn, Fe, Co, Ni, Cu, Zn, Mo, element with dual (essentiality/toxicity) behavior (Se), and in common toxic elements, such as As, Cd and Pb. Relative quantification of the element concentration was achieved by average signal normalization. Different elemental profiles were demonstrated for 51 out of 6000 mutagenized A. thaliana plants23 and for 212 out of 4385 investigated yeast mutant strains.78 The necessity for the global approach was corroborated by changes observed in levels of not only one but of multiple elements in most mutants.23,78 The observation that only 6 of the 50 Arabidopsis ion-profile mutants show changes in only one element was considered to strongly support the existence of regulatory networks in the ionome of plants.23
The precision of this approach as a functional genomics tool can be improved by integrating speciation-relevant information or by studies at cellular or subcellular resolution. In particular, the use of laser ablation sampling of XAS imaging holds promise for development of high-resolution metal imaging.55 Such technologies offer more precise approaches to metallomics, allowing changes in the total shoot, seed or root metallomes to be mapped. Metallomics imaging would also allow colocalization of in vivogene expression and protein localization patterns with the element concentration blueprint changes thus providing spatial linkage between gene, protein and metal function.78
7.2 Identification of metalloproteins in protein sets obtained by expression of individual genes
In this approach, schematically shown in Fig. 11, genes (open reading frames: portions of an organism’s genome that contains a sequence of bases that could potentially encode a protein) are cloned with purification tags, and recombinant proteins are expressed from suitable microbial hosts, e.g. E. coli, and purified by affinity chromatography.79,80 The approach leads to the availability of a relatively pure protein with the known primary sequence and is the basis of structural genomics (the high-throughput determination of the 3D structures of all proteins of a given organism, by e.g.X-ray crystallography, NMR spectroscopy or computational approaches).79,80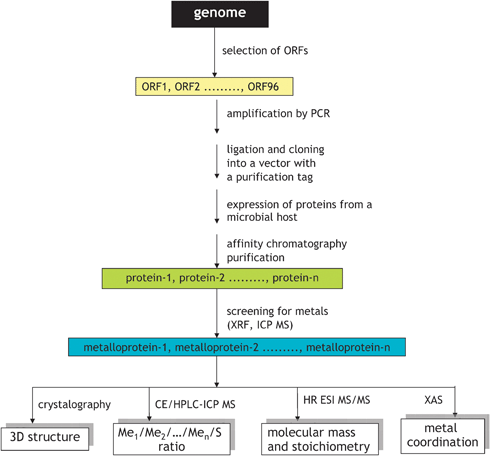 | ||
| Fig. 11 In vitro workflow for metalloproteomics based on structural genomics approach. Adapted from ref. 82. | ||
It is expected that proteins requiring a metal ion for their function (metalloproteins) will be purified and crystallized as metal–protein complexes.81–83 Hence, the multielement analysis of all the proteins resulted from a study allows the selection of proteins with the affinity to particular elements.82,83 Indeed, large scale studies of proteins, stoichiometrically binding Co, Cu, Ni and Zn79,82,83 but also Mn and Fe,79,83 were reported. The purity of the selected proteins is generally sufficient for XANES and EXAFS studies allowing the determination of the oxidation state and the description of the coordination center.84
In addition, the metal–ion binding stoichiometry, structure and dynamics can be investigated by electrospray MS,85,86 sometimes after on-line desalting by size-exclusion LC. High resolution separation techniques, e.g.capillary electrophoresis, coupled with multiisotopic detection by ICP MS are particularly useful for the termination of the metal-ion protein binding stoichiometry using a minute sample amount.87,88
The individual gene cloning followed by heterologous protein expression is an attractive experimental approach to the identification of metal-binding sequence motives in proteins. Assuming that such motives are conserved over various organisms, structures of metalloproteins can be predicted by a genome search using bioinformatics tools (cf. section 8). It is, however, unclear to what extent a protein obtained by expression of an individual gene retains biological relevance. The formation of a metal site often requires a set of post-translational events that may not take place in a simplified model system. For example, microbial hosts used for HT expression may not possess the specific chaperones needed by the metalloprotein for metal incorporation.81 Some metals can also be toxic to the host organism, which spurs interest in the use of cell-free protein expression systems.81 The fate of the metal during purification of the expressed proteins has never been monitored, so the metalloproteins found are only those that did not dissociate during the purification step. For these reasons, the HT cloning should be considered as complementary to the expression of the entire proteome under given environmental conditions and the in vivo analysis of metalloproteome, as discussed in section 5.
7.3 Correlating the metallometabolome with specific gene functions
Correlations between the genome and the metallometabolome are not so evident as between the genome and the metalloproteome, and may even be absent. A good example of a search for such interactions can be the search for genes conferring metal resistance to an organism, e.g. a hyperaccumulating plant, using molecular cloning by complementation and screening on a metal-rich toxic medium.89The approach consists of first creating a gene library (bank) of the organism under study (e.g. a plant).90 The library is referred to as a population of organisms (e.g. yeast cells) each of which carries a DNA fragment of the plant. The creation of the library includes the isolation of the DNA of an organism of interest, sometimes digestion to different lengths to ensure that all the genes have been digested to manageable sizes and ligation to vector plasmids to be taken up by the host (yeast). As a result, statistically all the genes will be incorporated into the plasmids, and consequently in the yeast (each gene in a different cell).
The following step is aimed at isolating cells containing a gene involved in resistance to the metal, which can be achieved by submitting the whole yeast population to a stress using a metal concentration considered as toxic to normal yeast cells. It is expected that only the cells containing the resistance gene will survive and continue to grow. The genome of the harvested yeast cells will be different from control yeast by the presence of the resistance gene; its proteome may contain proteins coded by this gene whereas the metabolome ligands are involved in the metal resistance.
A practical application of this principle is demonstrated in Fig. 12.89 A gene library of a model polymetal hyperaccumulating plant, Thlaspi caerulescens, was expressed into yeast producing ca. 400000 clones. Out of all of them only a few survived the toxicity test with the lethal nickel concentration and produced a population of Ni-resistant yeast cells. The Ni metabolome analysis by size-exclusion LC followed by capillary electrophoresis with the parallel ICP MS and ESI MS/MS detection showed a single Ni-complexing ligand that gave tandem mass spectra identical to a Ni-nicotianamine synthetic standard. The Ni-nicotianamine complex was found to be the only Ni-complex present in the wild-type plant. The DNA analysis of the yeast showed the presence of a gene with 98% homology to the gene coding for nicotianamine synthase, an enzyme previously reported to be involved in the metabolism and transport of iron.91
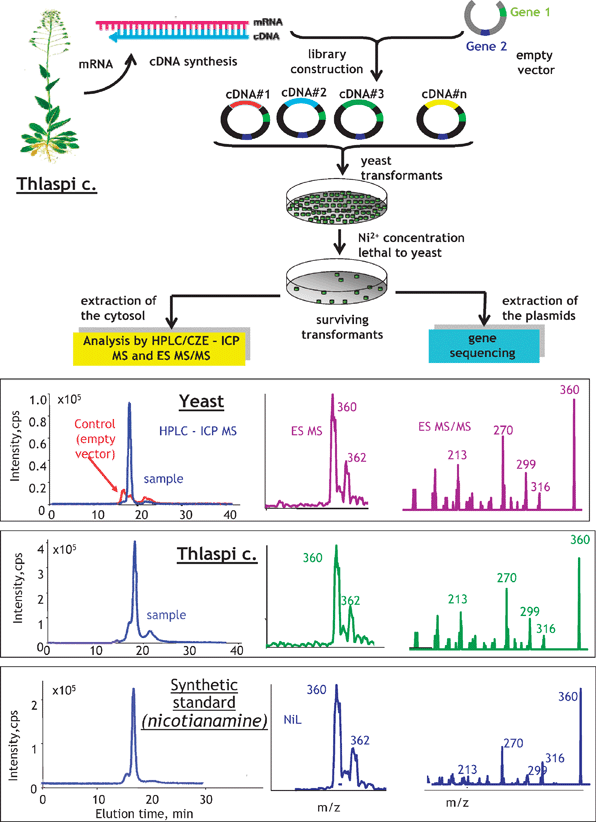 | ||
| Fig. 12 Search for a metal-relevant gene by a combination of mass spectrometric metallometabolite identification and molecular cloning by complementation on a toxic medium. The example concerns a search for a gene relevant to the Ni-transport in Thlaspi caerulescence. Adapted from ref. 89. | ||
The combination of metabolomics with the molecular cloning by the complementation approach offers the advantage of correlation of identified metabolites with functional genes present in the studied organism.
8. Bioinformatics: prediction of the metal-binding patterns
The availability of complete genomic sequences of an increasing number of organisms spurs interest in the development of bioinformatics tools allowing the prediction (in silico identification) of full sets of trace-element containing proteins.92,93A pioneering study reported the development of bioinformatics methods to identify almost all genes encoding selenocysteine-containing proteins in a number of organisms, including humans. The original computer algorithms were based on the use of the canonical SECIS (selenocysteine insertion sequence) element (AUGA_AA_GA) as a signature for mammalian selenoproteins94 and then refined to search for SECIS-containing genes with in-frame UGA codons.95SECIS element is harbored by selenoprotein mRNAs in their 3′ untranslated region and triggers the recoding of the UGA codon (normally stop codon) into selenocysteine. Note that whereas the eukaryotic SECIS element consensus is well characterized and can be identified in data bases, conservation of SECIS element in bacteria was insufficient for their computational description.96 Alternative SECIS-independent identification of selenocysteine–cysteine pairs was proposed.96
Methods allowing the search for entire sets of metal-binding proteins are less advanced. Bertini et al. proposed an approach taking advantage of known consensus sequences, i.e. taking into account the nature and spacing of amino acids present in the metal-binding region.93 The bioinformatic analysis of the protein sequence (by correlation with the relevant gene) allows the finding of the conserved metal-binding motifs and provides important clues to function and metal site structure.83 For example, the most abundant class of Zn-binding proteins in humans is that of zinc-fingers, with Cys4 and Cys2His2 as consensus sequence.10 This approach is critically dependent on the availability of consensus sequences for the binding of different metals. Some are present in databases, e.g. at Scripps Research Institute,97 and more are expected to be found by experimental structural genomics.
Bioinformatics is also useful for the prediction of a metal-binding site based on a known 3D structure. It is possible owing to the conserved nature of the metal-binding site and its usual compact size.98 The number of 3D protein structures is still limited but increases with the progress of structural genomics projects. Metal-binding sites can also be predicted by combination of low-resolution structural data with sequence information.99
The difficulties with the experimental methods for the characterization of seleno- and metalloproteomes spur interest in in silico approaches which, in some case, not only can facilitate the experimental research in metallomics (as the availability of genomics sequences revolutionized proteomics) but also can provide stand-alone insights into the occurrence and role of metalloproteins. Recently, a series of bioinformatics studies reported the distribution of iron-, copper- and Zn-binding proteins in several archaea, bacteria and eukaryotes.93
9. Quantitative metallomics
As quantification by molecular MS is not straightforward, the canonical proteomics approaches allow the qualitative (primary sequence identification) information only. Quantitative methods in proteomics are based on the use of protein- (or protein class-) specific isotopic tags and differential quantification between the control and the analyzed cell- or organism state.100,101 Quantitative metabolomics is based on the derivatization of the target analytes to confer them suitable UV-VIS absorption or fluorescence characteristics followed by quantification using a calibration (or standard addition) curve established with the corresponding standards. These methods ignore the presence of the metal and address the quantification of the ligand only.The heteroatom present in the species constituting the metallome or metalloproteome can be used as a tag for their quantification,102 usually by ICP MS, owing to its isotopic specificity, high elemental selectivity regardless of the metal-coordination environment and the sample matrix, the dynamic range of at least 6 decades, and the ease of coupling with separation techniques.103
In the simplest case, ICP MS can be used as a multielement analytical technique to provide a multielement blueprint of a tissue to be correlated with is genetic profile.23,78 Such a blueprint can be obtained with 50 μm (ICP MS) or submicrometre (XAFS) resolution providing a metallomics localization pattern.55
Species-specific quantification requires chromatographic or electrophoretic resolution prior to ICP MS. The mandatory condition is the chromatographic (electrophoretic) purity of the analyte (i.e. the peak should not contain but a single species of a given element). The methods of quantification include:
(i) Chromatographic (electrophoretic) quantification using the external or standard addition calibration. This approach requires the availability of a well characterized standard. Examples include the quantification of ceruloplasmin (Cu),104myoglobin (Fe)105 or some selenoproteins.106
(ii) Quantification by species-specific isotope dilution. A standard of the isotopically enriched analyte must be available. The standard must not exchange the isotope with the environment during the procedure. This condition is naturally fulfilled in the case of a covalently incorporated heteroelement, e.g.77Se-labelled selenomethionine107 or Asp-Tyr-77SeMet-Gly-Ala-Ala-Lys peptide,108 but also for a number of metalloproteins (57Fe-transferrin,10965Cu-rustycyanin110 or 57Fe-ferritin111),
(iii) quantification by species-unspecific isotope dilution.112
The approach is schematically shown in Fig. 13. The effluent from an HPLC column (Fig. 13a) or from an electrophoretic capillary (Fig. 13b) is mixed with a solution containing an enriched isotope of the analyte element. The latter serves as an internal standard correcting for the nebulization and ionization efficiency. The method is based on the assumption of the identical element response from the sample, which in some cases may not be true. The validity of the measurement is critically dependent on the quantitative recovery of the analyte from the column, which is unfortunately seldom checked. The method, theoretically, does not require the knowledge of the analyte’s identity and is often used for the determination of the mass balance of the different species of a given element.113 Self-evident conditions are the existence of at least two stable isotopes of the analyte element and the absence of isobaric interferences for both of them. The typical applications include the quantification of isoforms of metallothionein,112 transferrin109 or haemoglobin114 and selenoproteins.115
 | ||
| Fig. 13 Quantitative determination of metal(loid) species by species-unspecific post-column isotopic dilution. (a) Scheme of an HPLC-ICP MS experimental setup; (b) scheme of a capillary electrophoresis LC-ICP MS experimental setup; (c) determination of the tryptic digestion efficiency of Se–calmoduline by nanoHPLC-ICP MS;113 (d) determination of the Cd-metallothionein stoichiometry by capillary electrophoresis-ICP MS.87 | ||
A practical illustration of the approach is shown in Fig. 13c for the determination of the digestion efficiency of selenocalmoduline.113 The chromatogram allows the quantification of individual peptides (provided they contain a heteroelement), of the partly digested protein and of the non-digested protein. In metalloproteomics, the approach can be useful for the determination of the metal–protein binding stoichiometry (Fig. 13d). If a metalloprotein contains at least one sulfur atom (methionine or cysteine) and leaves the column chromatographically pure, the ratio of the complexed metal to sulfur can be determined thus producing the empiric formula of the complex. In this way, the stoichiometry of the Cd-MT complexes with the different metallothionein isoforms in rat kidney could be determined.87
Note the increasing popularity of methods based on the derivatization (external tagging) by saturation with a metal or by reaction with a metal-containing reagent.100 These methods are not considered metallomics methods as the analyte is not a metallospecies.
10. Conclusions
The understanding of interactions and functional connections of metal ions and their species with genes, proteins, metabolites and other biomolecules within organisms and ecosystems is critically dependent on the knowledge of the entirety of metal and metalloid species present in a cell or tissue type, their identity, quantity and localization. This is becoming increasingly possible owing to the progress in in vivo analytical methods, in vitro functional analysis and in silico analysis using bioinformatics. The emerging field of metallomics addresses the systematic view of the methodology, role and function of trace elements across several disciplines including biological catalysis, biogeochemistry and environmental chemistry, clinical chemistry, plant biochemistry and physiology, and nutrition and essential elements supplementation.References
- R. D. Fleischmann, M. D. Adams, O. White, R. A. Clayton, E. F. Kirkness, A. R. Kerlavage, C. J. Bult, J.-F. Tomb, B. A. Dougherty, J. M. Merrick, K. McKenney, G. Sutton, W. FitzHugh, C. Fields, J. D. Gocayne, J. Scott, R. Shirley, L.-I. Liu and J. C. Venter, Science, 1995, 269, 496 CAS.
- International Human Genome Sequencing Consortium, Nature, 2001, 409, 860 CrossRef CAS.
- J. Craig Venter, M. D. Adams, E. W. Myers, P. W. Li, R. J. Mural, G. G. Sutton, H. O. Smith, M. Yandell, C. A. Evans, R. A. Holt, J. D. Gocayne, P. Amanatides, R. M. Ballew, D. H. Huson, J. R. Wortman, Q. Zhang, C. D. Kodira, X. H. Zheng, L. Chen, M. Skupski, G. Subramanian, P. D. Thomas, J. Zhang, G. L. Gabor Miklos, C. Nelson, S. Broder, A. G. Clark, J. Nadeau, V. A. McKusick, N. Zinder, A. J. Levine, R. J. Roberts, M. Simon, C. Slayman, M. Hunkapiller, R. Bolanos, A. Delcher, I. Dew, D. Fasulo, M. Flanigan, L. Florea, A. Halpern, S. Hannenhalli, S. Kravitz, S. Levy, C. Mobarry, K. Reinert, K. Remington, J. Abu-Threideh, E. Beasley, K. Biddick, V. Bonazzi, R. Brandon, M. Cargill, I. Chandramouliswaran, R. Charlab, K. Chaturvedi, Z. Deng, V. Di Francesco, P. Dunn, K. Eilbeck, C. Evangelista, A. E. Gabrielian, W. Gan, W. Ge, F. Gong, Z. Gu, P. Guan, T. J. Heiman, M. E. Higgins, R.-R. Ji, Z. Ke, K. A. Ketchum, Z. Lai, Y. Lei, Z. Li, J. Li, Y. Liang, X. Lin, F. Lu, G. V. Merkulov, N. Milshina, H. M. Moore, A. K. Naik, V. A. Narayan, B. Neelam, D. Nusskern, D. B. Rusch, S. Salzberg, W. Shao, B. Shue, J. Sun, Z. Y. Wang, A. Wang, X. Wang, J. Wang, M.-H. Wei, R. Wides and C. Xiao, Science, 2001, 291, 1304 CrossRef CAS.
- W. P. Blackstock and M. P. Weir, Trends Biotechnol., 1999, 17, 121 CrossRef CAS.
- N. L. Anderson and N. G. Anderson, Electrophoresis, 1998, 19, 1853 CrossRef CAS.
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64 CrossRef CAS.
- M. Karas and F. Hillenkamp, Anal. Chem., 1988, 60, 2299 CrossRef CAS.
- K. Tanaka, H. Waki, Y. Ido, S. Akita, Y. Yoshida and T. Yoshida, Rapid Commun. Mass Spectrom., 1988, 2, 151 CAS.
- J. A. Tainer, V. A. Roberts and E. D. Getzoff, Curr. Opin. Biotechnol., 1991, 2, 582 CrossRef CAS.
- W. Maret, J. Anal. At. Spectrom., 2004, 19, 15 RSC.
- B. P. Rosen, Top. Curr. Genet., 2006, 14, 485–505 Search PubMed.
- A. C. Rosenzweig, Chem. Biol., 2002, 9, 673 CrossRef CAS.
- C. E. Outten and T. V. O'Halloran, Science, 2001, 292, 2488 CrossRef CAS.
- D. J. Thiele and J. D. Gitlin, Nat. Chem. Biol., 2008, 4, 145 CrossRef CAS.
- R. J. P. Williams, Coord. Chem. Rev., 2001, 216–217, 583 CrossRef CAS.
- J. Szpunar, Anal. Bioanal. Chem., 2004, 378, 54 CAS.
- H. Haraguchi, J. Anal. At. Spectrom., 2004, 19, 5 RSC.
- J. Szpunar, Analyst, 2005, 130, 442 RSC.
- J. Lopez-Barea and J. L. Gomez-Ariza, Proteomics, 2006, 6, S51 CrossRef.
- W. Shi and M. R. Chance, Cell. Mol. Life Sci., 2008, 65, 3040 CrossRef CAS.
- N. Jakubowski, R. Lobinski and L. Moens, J. Anal. At. Spectrom., 2004, 19, 1 RSC.
- D. W. Koppenaal and G. M. Hieftje, J. Anal. At. Spectrom., 2007, 22, 855 RSC.
- B. Lahner, J. Gong, M. Mahmoudian, E. L. Smith, K. B. Abid, E. E. Rogers, M. L. Guerinot, J. F. Harper, J. M. Ward, L. McIntyre, J. I. Schroederk and D. E. Salt, Nat. Biotechnol., 2003, 21, 1215 CrossRef CAS.
- A. Sanz-Medel, Anal. Bioanal. Chem., 2005, 381, 1 CrossRef CAS.
- N. Jakubowski and G. M. Hieftje, J. Anal. At. Spectrom., 2007, 23, 13 Search PubMed.
- C. Andreini, I. Bertini, G. Cavallaro, G. L. Holliday and J. M. Thornton, JBIC, J. Biol. Inorg. Chem., 2008, 13, 1205 CrossRef CAS.
- R. J. P. Williams and J. J. R. Frausto Da Silva, J. Chem. Educ., 2004, 81, 738 CrossRef CAS.
- A. L. Zerkle, C. H. House and S. L. Brantley, Am. J. Sci., 2005, 305, 467 Search PubMed.
- C. L. Dupont, S. Yang, B. Palenik and P. E. Bourne, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17822 CrossRef CAS.
- J. E. Mayer, W. H. Pfeiffer and P. Beyer, Curr. Opin. Plant Biol., 2008, 11, 166 CrossRef CAS.
- K. Shah and J. M. Nongkynrih, Biol. Plant., 2007, 51, 618 CrossRef CAS.
- S. Clemens, Planta, 2001, 212, 475 CrossRef CAS.
- P. P. Kulkarni, Y. M. She, S. D. Smith, E. A. Roberts and B. Sarkar, Chem.–Eur. J., 2006, 12, 2410 CrossRef CAS.
- H. J. Thierse, S. Helm and P. Pankert, Methods Mol. Biol. (Clifton, N. J.), 2008, 425, 139 Search PubMed.
- V. Lopez-Avila and J. V. Spencer, Clin. Med. Cardiol., 2008, 2, 75 Search PubMed.
- Z. Guo and P. J. Sadler, Angew. Chem., Int. Ed., 1999, 38, 1513.
- G. N. Schrauzer, J. Nutr., 2000, 130, 1653 CAS.
- M. P. Rayman, Lancet, 2000, 356, 233 CrossRef CAS.
- S. Beranova-Giorgianni, Trends Anal. Chem., 2003, 22, 273 CrossRef CAS.
- E. Nagele, M. Vollmer, P. Horth and C. Vad, Expert Rev. Proteomics, 2004, 1, 37 Search PubMed.
- N. L. Kelleher, Anal. Chem., 2004, 76, 197A.
- F. W. McLafferty, K. Breuker, M. Jin, X. Han, G. Infusini, H. Jiang, X. Kong and T. P. Begley, FEBS J., 2007, 274, 6256 CAS.
- P. Giusti, D. Schaumloffel, H. Preud'homme, J. Szpunar and R. Lobinski, J. Anal. At. Spectrom., 2006, 21, 26 RSC.
- M. Dernovics, P. Giusti and R. Lobinski, J. Anal. At. Spectrom., 2007, 22, 41 RSC.
- H. Roelofsen, R. Balgobind and R. J. Vonk, J. Cell. Biochem., 2004, 93, 732 CrossRef CAS.
- Y. M. She, S. Narindrasorasak, S. Yang, N. Spitale, E. A. Roberts and B. Sarkar, Mol. Cell. Proteomics, 2003, 2, 1306 CrossRef CAS.
- S. D. Smith, Y.-M. She, E. A. Roberts and B. Sarkar, J. Proteome Res., 2004, 3, 834 CrossRef CAS.
- C. Basset, A. Dedieu, P. Guerin, E. Quemeneur, D. Meyer and C. Vidaud, J. Chromatogr., A, 2008, 1185, 233 CrossRef CAS.
- R. Ge, X. Sun, Q. Gu, R. M. Watt, J. A. Tanner, B. C. Y. Wong, H. H. Xia, J.-D. Huang, Q.-Y. He and H. Sun, JBIC, J. Biol. Inorg. Chem., 2007, 12, 831 CrossRef CAS.
- K. Heiss, C. Junkes, N. Guerreiro, M. Swamy, M. M. Camacho-Carvajal, W. W. A. Schamel, I. D. Haidl, D. Wild, H. U. Weltzien and H.-J. Thierse, Proteomics, 2005, 5, 3614 CrossRef CAS.
- C. C. S. Kung, W. N. Huang, Y. C. Huang and K. C. Yeh, Proteomics, 2006, 6, 2746 CrossRef CAS.
- I. Wittig, H.-P. Braun and H. Schagger, Nat. Protocols, 2006, 1, 418 CAS.
- J. L. Neilsen, A. Abildtrup, J. Christensen, P. Watson, A. Cox and C. W. McLeod, Spectrochim. Acta, Part B, 1998, 53, 339 CrossRef.
- R. Ma, C. W. McLeod, K. Tomlinson and R. K. Poole, Electrophoresis, 2004, 25, 2469 CrossRef CAS.
- R. Lobinski, C. Moulin and R. Ortega, Biochimie, 2006, 88, 1591 CrossRef CAS.
- J. S. Becker, M. Zoriy, J. S. Becker, C. Pickhardt and M. Przybylski, J. Anal. At. Spectrom., 2004, 19, 149 RSC.
- J. Su. Becker, M. V. Zoriy, J. Sa. Becker and R. Lobinski, Metallomics Search PubMed , submitted.
- L. Tastet, D. Schaumloffel and R. Lobinski, J. Anal. At. Spectrom., 2008, 23, 309 RSC.
- S. Chevreux, S. Roudeau, A. Fraysse, A. Carmona, G. Deves, P. L. Solari, T. C. Weng and R. Ortega, J. Anal. At. Spectrom., 2008, 23, 1117 RSC.
- J. Szpunar, Analyst, 2000, 125, 963 RSC.
- J. Szpunar and R. Lobinski, Anal. Bioanal. Chem., 2002, 373, 404 CrossRef CAS.
- J. L. Gardea-Torresdey, J. R. Peralta-Videa, G. De La Rosa and J. G. Parsons, Coord. Chem. Rev., 2005, 249, 1797 CrossRef CAS.
- J. Kawai, Absorption techniques in X-ray spectrometry, in Encyclopedia of Analytical Chemistry, ed. R. A. Meyers, Wiley, Chichester, 2000 Search PubMed.
- R. Terzano, Z. Al Chami, B. Vekemans, K. Janssens, T. Miano and P. Ruggiero, J. Agric. Food Chem., 2008, 56, 3222 CrossRef CAS.
- E. Montarges-Pelletier, V. Chardot, G. Echevarria, L. J. Michot, A. Bauer and J. L. Morel, Phytochemistry, 2008, 69, 1695 CrossRef CAS.
- K. Bluemlein, A. Raab, A. A. Meharg, J. M. Charnock and J. Feldmann, Anal. Bioanal. Chem., 2008, 390, 1739 CrossRef CAS.
- X. Feng and M. M. Siegel, Anal. Bioanal. Chem., 2007, 389, 1341 CrossRef CAS.
- M. Dernovics and R. Lobinski, Anal. Chem., 2008, 80, 3975 CrossRef CAS.
- G. Weber, N. von Wiren and H. Hayen, J. Sep. Sci., 2008, 31, 1615 CrossRef CAS.
- Y. Xuan, E. B. Scheuermann, A. R. Meda, H. Hayen, N. von Wiren and G. Weber, J. Chromatogr., A, 2006, 1136, 73 CrossRef CAS.
- T. Y. Yen, J. A. Villa and J. G. DeWitt, J. Mass Spectrom., 1999, 34, 930 CrossRef CAS.
- H. G. Infante, G. O’Connor, M. Rayman, R. Hearn and K. Cook, J. Anal. At. Spectrom., 2006, 21, 1256 RSC.
- L. Ouerdane, S. Mari, P. Czernic, M. Lebrun and R. Lobinski, J. Anal. At. Spectrom., 2006, 21, 676 RSC.
- A. J. Alpert, J. Chromatogr., A, 1990, 499, 177 CrossRef CAS.
- C. Casiot, V. Vacchina, H. Chassaigne, J. Szpunar, M. Potin-Gautier and R. Lobinski, Anal. Commun., 1999, 36, 77 RSC.
- P. A. Rea, Nat. Biotechnol., 2003, 21, 1149 CrossRef CAS.
- D. E. Salt, I. Baxter and B. Lahner, Annu. Rev. Plant Biol., 2008, 59, 709 CrossRef CAS.
- D. J. Eide, S. Clark, T. M. Nair, M. Gehl, M. Gribskov, M. L. Guerinot and J. F. Harper, Genome Biol., 2005, 6, R77 CrossRef.
- M. R. Chance, A. Fiser, A. Sali, U. Pieper, N. Eswar, G. Xu, J. E. Fajardo, T. Radhakannan and N. Marinkovic, Genome Res., 2004, 14, 2145 CrossRef CAS.
- M. R. Chance, A. R. Bresnick, S. K. Burley, J. S. Jiang, C. D. Lima, A. Sali, S. C. Almo, J. B. Bonanno, J. A. Buglino, S. Boulton, H. Chen, N. Eswar, G. He, R. Huang, V. Ilyin, L. McMahan, U. Pieper, S. Ray, M. Vidal and L. K. Wang, Protein Sci., 2002, 11, 723 CrossRef CAS.
- J. F. Hall, M. J. Ellis, T. Kigawa, T. Yabuki, T. Matsuda, E. Seki, S. S. Hasnain and S. Yokoyama, J. Synchrotron Radiat., 2005, 12, 4 CAS.
- R. A. Scott, J. E. Shokes, N. J. Cosper, F. E. Jenney and M. W. W. Adams, J. Synchrotron Radiat., 2005, 12, 19 CAS.
- W. Shi, C. Zhan, A. Ignatov, B. A. Manjasetty, N. Marinkovic, M. Sullivan, R. Huang and M. R. Chance, Structure, 2005, 13, 1473 CrossRef CAS.
- S. S. Hasnain, J. Synchrotron Radiat., 2004, 11, 7 CAS.
- H. Chassaigne, V. Vacchina and R. Lobinski, Trends Anal. Chem., 2000, 19, 300 CrossRef CAS.
- I. A. Kaltashov, M. Zhang, S. J. Eyles and R. R. Abzalimov, Anal. Bioanal. Chem., 2006, 386, 472 CrossRef CAS.
- K. Polec-Pawlak, D. Schaumlöffel, J. Szpunar, A. Prange and R. Lobinski, J. Anal. At. Spectrom., 2002, 17, 908 RSC.
- A. Prange and D. Profrock, Anal. Bioanal. Chem., 2005, 383, 372 CrossRef CAS.
- V. Vacchina, S. Mari, P. Czernic, L. Marques, K. Pianelli, D. Schaumloffel, M. Lebrun and R. Lobinski, Anal. Chem., 2003, 75, 2740 CrossRef CAS.
- M. D. Rose and J. R. Broach, Methods Enzymol., 1991, 194, 195 CAS.
- D. Mizuno, K. Higuchi, T. Sakamoto, H. Nakanishi, S. Mori and N. K. Nishizawa, Plant Physiol., 2003, 132, 1989 CrossRef CAS.
- V. N. Gladyshev, G. V. Kryukov, D. E. Fomenko and D. L. Hatfield, Annu. Rev. Nutr., 2004, 24, 579 CrossRef CAS.
- I. Bertini and A. Rosato, Eur. J. Inorg. Chem., 2007, 2546 CrossRef CAS.
- G. V. Kryukov, V. M. Kryukov and V. N. Gladyshev, J. Biol. Chem., 1999, 274, 33888 CrossRef CAS.
- S. Castellano, N. Morozova, M. Morey, M. J. Berry, F. Serras, M. Corominas and R. Guigo, EMBO Rep., 2001, 2, 697 CrossRef CAS.
- G. V. Kryukov and V. N. Gladyshev, EMBO Rep., 2004, 5, 538 CrossRef CAS.
- J. M. Castagnetto, S. W. Hennessy, V. A. Roberts, E. D. Getzoff, J. A. Tainer and M. E. Pique, Nucleic Acids Res., 2002, 30, 379 CrossRef CAS.
- J. W. H. Schymkowitz, F. Rousseau, I. C. Martins, J. Ferkinghoff-Borg, F. Stricher and L. Serrano, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10147 CrossRef CAS.
- J. S. Sodhi, K. Bryson, L. J. McGuffin, J. J. Ward, L. Wernisch and D. T. Jones, J. Mol. Biol., 2004, 342, 307 CrossRef CAS.
- S. E. Ong and M. Mann, Nat. Chem. Biol., 2005, 1, 252 CrossRef CAS.
- M. Bantscheff, M. Schirle, G. Sweetman, J. Rick and B. Kuster, Anal. Bioanal. Chem., 2007, 389, 1017 CrossRef CAS.
- A. Sanz-Medel, Anal. Bioanal. Chem., 2008, 391, 885 CrossRef CAS.
- A. A. Ammann, J. Mass Spectrom., 2007, 42, 419 CrossRef CAS.
- V. Lopez-Avila, O. Sharpe and W. H. Robinson, Anal. Bioanal. Chem., 2006, 386, 180 CrossRef CAS.
- C. F. Harrington, S. Elahi, S. A. Merson and P. Ponnampalavanar, J. AOAC Int., 2004, 87, 253 CAS.
- P. Jitaru, G. Cozzi, A. Gambaro, P. Cesco and C. Barbante, Anal. Bioanal. Chem., 2008, 391, 661 CrossRef CAS.
- J. R. Encinar, D. Schaumloffel, Y. Ogra and R. Lobinski, Anal. Chem., 2004, 76, 6635 CrossRef CAS.
- A. Polatajko, J. R. Encinar, D. Schaumloffel and J. Szpunar, Chem. Anal. (Warsaw), 2005, 50, 265 CAS.
- M. E. Del Castillo Busto, M. Montes-Bayon and A. Sanz-Medel, Anal. Chem., 2006, 78, 8218 CrossRef CAS.
- C. F. Harrington, D. S. Vidler, M. J. Watts and J. F. Hall, Anal. Chem., 2005, 77, 4034 CrossRef CAS.
- M. Hoppler, L. Meile and T. Walczyk, Anal. Bioanal. Chem., 2008, 390, 53 CrossRef CAS.
- D. Schaumlöffel and R. Lobinski, Int. J. Mass Spectrom., 2005, 242, 217 Search PubMed.
- P. Giusti, D. Schaumloffel, J. R. Encinar and J. Szpunar, J. Anal. At. Spectrom., 2005, 20, 1101 RSC.
- M. E. Del Castillo Busto, M. Montes-Bayon, E. Anon and A. Sanz-Medel, J. Anal. At. Spectrom., 2008, 23, 758 RSC.
- L. Hinojosa Reyes, J. M. Marchante-Gayon, J. I. Garcia Alonso and A. Sanz-Medel, J. Anal. At. Spectrom., 2003, 18, 1210 RSC.
| This journal is © The Royal Society of Chemistry 2009 |