 Open Access Article
Open Access ArticleAn efficient method for synthesising unsymmetrical silaketals: substrates for ring-closing, including macrocycle-closing, metathesis†
Christopher
Cordier
ab,
Daniel
Morton
ab,
Stuart
Leach
ab,
Thomas
Woodhall
ab,
Catherine
O'Leary-Steele
ab,
Stuart
Warriner
ab and
Adam
Nelson
*ab
aSchool of Chemistry, University of Leeds, Leeds, UK LS2 9JT. E-mail: a.s.nelson@leeds.ac.uk; Fax: +44 (0)113 343 6565; Tel: +44 (0)113 343 6502
bAstbury Centre for Structural Molecular Biology, University of Leeds, Leeds, UK LS2 9JT
First published on 10th April 2008
Abstract
Diisopropylsilyl ethers were activated with N-bromosuccinimide, and reacted with a fluorous-tagged alcohol, to yield tethered substrates for ring-closing metathesis reactions.
The use of temporary tethers between reactants is an extremely valuable approach in synthetic organic chemistry.1 The approach can promote selective reaction between the tethered components, often under much milder conditions than the corresponding intermolecular process. Furthermore, reactions between the tethered components often proceed with stereoselectivities that complement, or improve on, those observed in intermolecular reactions.
Silicon tethers can control the cross-metathesis of pairs of allylic or homoallylic alcohols (e.g.1 and 2, Scheme 1). After tethering of the reactants (e.g. → 3), a ring-closing metathesis reaction gives a cyclic silaketal; silaketal cleavage (e.g.4 → 5) then yields the Z isomer of net cross-metathesis of the components. The approach has been used in target2 and library3 synthesis, for example in the synthesis of attenol A,2amucocin,2b and the epothilones.2c Furthermore, tethering metathesis reactions can lead to long-range diastereoselection.4
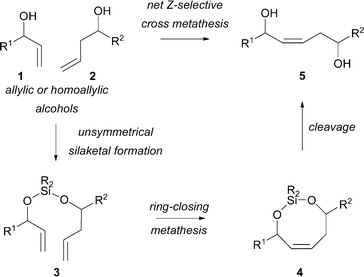 | ||
| Scheme 1 Net Z-selective cross-metathesis of an allylic and a homoallylic alcohol. | ||
A number of methods2,4,5 have been developed for the selective synthesis of the required unsymmetrical silaketals. Usually, one of the alcohols is reacted with an excess of a volatile dichlorosilane.2a–b,4 Alternatively, reaction of the anion of one of the alcohols with a dichlorosilane can give the monosubstituted reagent needed to prepare selectively the unsymmetrical product.2c In this communication, we describe an improved efficient and selective method for preparing unsymmetrical silaketal substrates for ring-closing (including macrocycle-closing) metathesis reactions.
We required an efficient method for the formation of unsymmetrical silaketals from the fluorous-tagged alcohol66 (Scheme 2). The key design features of the fluorous-tagged linker 6 are summarised in Scheme 3. A fluorous tag was incorporated to facilitate the purification by fluorous-solid phase extraction7 (F-SPE) of the required silaketals 7 from excess reagents used. The linker 6 was designed6 such that metathesis of the unsymmetrical silaketals (e.g.7d) would release the required cyclic products (e.g.9) from the fluorous tag, thus facilitating purification by F-SPE. Subsequent cyclisation of 10 (→ 11) would then regenerate the catalytically active methylene complex.8
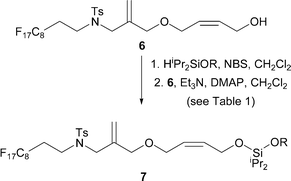 | ||
| Scheme 2 Synthesis of fluorous-tagged unsymmetrical silaketals 7a–l. See Fig. 1 for definitions of the R groups. | ||
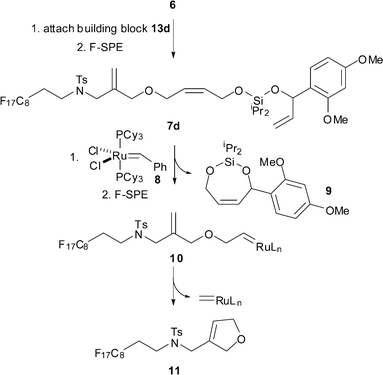 | ||
| Scheme 3 Design of the fluorous-tagged linker 6 illustrated for the proposed synthesis of the cyclic silaketal 9. | ||
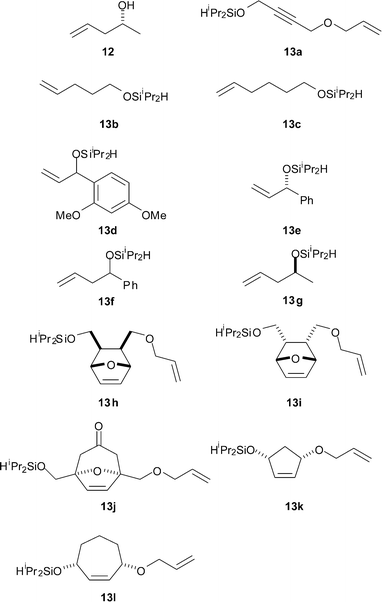 | ||
| Fig. 1 Substrates, including the diisopropylsilyl ethers HiPr2SiOR (13), used in this study. | ||
To start with, we investigated Malacria's method for the formation and activation of diisopropylsilyl ethers (entry 1, Table 1).9 Hence the alcohol 12 was silylated, activated with NBS, and the resulting bromosilane reacted with the fluorous-tagged alcohol 6. Unfortunately, only a trace of the required silaketal was isolated, and the fluorous-tagged alcohol was recovered in >98% yield.
| Entry | Substrate | Methoda | Product | Yield (and purity) after F-SPE only (%)b | Yield of purified product (%)c |
|---|---|---|---|---|---|
| a Methods: A: (i) 12 (0.5 M), iPr2HSiCl (1 eq.), Et3N (1.1 eq.), DMAP (0.1 eq.), CH2Cl2, 0 °C → rt; (ii) NBS (1 eq.); (iii) 6 (1 eq.), Et3N (1.1 eq.), DMAP (0.1 eq.), CH2Cl2, 0 °C → rt; B: (i) 13 (3.5 eq., 0.2 M), NBS (3 eq.), CH2Cl2, 0 °C → rt, 15 min; (ii) add to 6 (1 eq., 40 mM), Et3N (15 eq.), DMAP (10 mol%), CH2Cl2, 0 °C → rt; (iii) F-SPE; C: (i) 13 (3.5 eq., 0.2 M), NBS (3 eq.), CH2Cl2, 0 °C → rt, 15 min; (ii) inverse addition of 6 (1 eq., 0.25 M), DMAP (50 mol%), Et3N, 0 °C → rt; (iii) F-SPE; D: (i) 13 (5.5 eq., 0.2 M), NBS (5 eq.), CH2Cl2, 0 °C → rt; (ii) inverse addition of 6 (1 eq., 0.25 M), DMAP (50 mol%), Et3N, 0 °C → rt; (iii) F-SPE. b Yield of product after F-SPE; the purity, determined by analytical HPLC, is given in parentheses. c Yield of product after purification on Florisil®. d An alcohol was used as the substrate; method A involves the synthesis of the diisopropylsilyl etherin situ. e The fluorous-tagged alcohol 6 was recovered in >98% yield. f In the absence of DMAP, an 86% yield of material with 56% purity was obtained. g Determined by 500 MHz 1H NMR spectroscopy. | |||||
| 1 | 12 d | A | ent-7g | — | Tracee |
| 2a | 13a | B | 7a | 98 (65)f | — |
| 2b | rac-13d | B | rac-7d | — | 56 |
| 3a | 13a | C | 7a | 94 (98) | — |
| 3b | 13b | C | 7b | >98 (98) | — |
| 3c | 13c | C | 7c | >98 (98) | — |
| 3d | rac-13d | C | rac-7d | — | 56 |
| 3e | 13e | C | 7e | 55 (93) | — |
| 3f | rac-13f | D | rac-7f | >98 (76) | — |
| 3g | 13g | C | 7g | 96 (85) | — |
| 3h | rac-13h | C | rac-7h | — | 54 |
| 3i | rac-13i | C | rac-7i | 93 (67) | 59 |
| 3j | rac-13j | C | rac-7j | — | 32 |
| 3k | rac-13k | C | rac-7k | — | 64 |
| 3l | 13l | C | 7l | 95 (>85g) | — |
We therefore re-investigated the activation of the racemic diisopropylsilyl etherrac-13d with NBS. NBS was added to a sample of the silyl ether 13d in CDCl3, and the reaction was monitored by 500 MHz 1H NMR spectroscopy (see Fig. 2). Within 15 min, the silyl ether 13d had been consumed, and had been converted cleanly into the corresponding bromosilane.
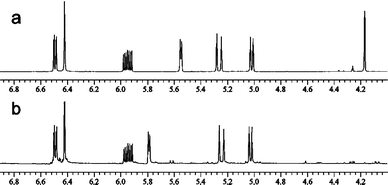 | ||
| Fig. 2 Activation of the diisopropylsilyl etherrac-13d. 500 MHz 1H NMR spectra of the silyl ether 13d in CDCl3 (a) before treatment with NBS (SiH: δ = 4.2 ppm); and (b) 15 min after treatment with NBS. | ||
With a method for the activation of a diisopropylsilyl ether in hand, we investigated the formation of unsymmetrical silaketals from the silyl ethers 13a and 13d (entries 2a–b, Table 1). The silyl ethers (3.5 eq., 0.2 M in CH2Cl2) were treated with NBS (3 eq.) and, after 15 min, were added to a solution of the fluorous-tagged alcohol 6 (1 eq., 40 mM) and DMAP (10 mol%) in CH2Cl2. Unfortunately, neither of the reactions proceeded to completion: in each case, following F-SPE, both the fluorous-tagged alcohol 6 and the required silaketal 7 were found in the fluorous fraction. Although the silaketals could be purified by filtration through a short pad of Florisil®, the method was not sufficiently efficient for application in parallel synthesis.
We increased the efficiency of the transformation by developing a protocol in which the concentration of the fluorous-tagged alcohol 6 was higher (entries 3a–l, Table 1). The diisopropylsilyl ethers 13a–l were prepared by silylation of the corresponding alcohols, and were used without further purification. The modified procedure involved inverse addition of a solution of the fluorous-tagged alcohol 6 and DMAP in triethylamine to a solution of the activated silyl ether in CH2Cl2. Following evaporation, addition of pentane and filtration, the crude reaction mixtures were dissolved in DMF, purified by F-SPE, and the fluorous fractions analysed, usually by HPLC. In some cases, the unsymmetrical silaketal products 7 were purified by filtration through a Florisil® pad.
The reactions of the silyl ethers 13a–c, derived from unhindered primary alcohols, were highly efficient: the unsymmetrical silaketals 7a–c were obtained in high yield (entries 3a–c, Table 1). With the silyl ethers 13d–g, derived from secondary alcohols, good yields of the corresponding silaketals 7d–g were obtained (entries 3d–f). We investigated more hindered bridged (13h–j) and cyclic substrates (13k–l) (entries 3h–l). Even when the reaction was not highly efficient, reasonable yields of the required silaketals were obtained after filtration through a Florisil® pad.
The chemoselectivity of our method for the activation of diisopropylsilyl ethers is remarkable. The silyl ethers 13a–l contain a range of functional groups that may react with NBS: alkenes, including strained substrates (13h–i); electron-rich aromatic rings (13d) and an alkyne (13a). We never observed any brominated products.
The ring-closing metatheses of five unsymmetrical fluorous-tagged silaketals (7d, 7f, 7h, 7j and 7l) were investigated (Table 2). The silaketals 7 were treated with 3 mol% portions of Grubbs’ first generation catalyst (8) until the reactions were judged to be complete by TLC. Work-up involved treatment with P(CH2OH)3, Et3N and silica, filtration through Celite,10 and evaporation. The products of the metathesis reactions described in Table 2 are shown in Fig. 3.
| Entry | Substrate | Methoda | Catalyst b (mol%) | Time/days | Product (yieldc) |
|---|---|---|---|---|---|
| a Methods: A: (i) 7 (1 mM), CH2Cl2, 45 °C; (ii) P(CH2OH)3 then Et3N; (iii) F-SPE; B: (i) 7 (1 mM), CH2Cl2, 45 °C; (ii) P(CH2OH)3 then Et3N; (iii) F-SPE; (iv) HF·pyridine then Me3SiOMe. b The catalyst was added at regular intervals in 3 mol% portions. c Yield of products (see Fig. 3) purified by column chromatography. d The product was purified by preparative HPLC. e 55 : 45 E : Z isomers mixture. | |||||
| 1 | 7d | A | 8 (3 × 3) | 0.3 | 9 (71%) |
| 2 | 7f | B | 8 (5 × 3) | 7 | 14 (19%) |
| 3a | 7h | A | 8 (4 × 3) | 1.1 | 15 (8%), 16 (23%)d |
| 3b | 7h | B | 8 (4 × 3) | 1 | 15 (21%)d |
| 4 | 7j | B | 8 (2 × 3) | 0.3 | 17 (35%)d,e |
| 5 | 7l | B | 8 (8 × 3) | 13 | 18 (21%) |
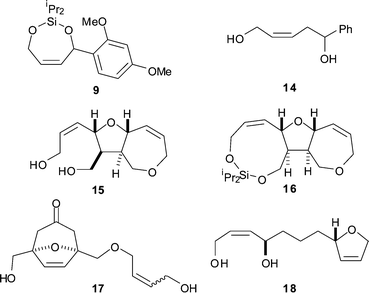 | ||
| Fig. 3 Products of the ring-closing metathesis reactions. | ||
Treatment of the silaketal 7d with the catalyst 8 triggered the release of the stable cyclic silaketal 9 from the fluorous tag (entry 1, Table 2). In the case of 7h, however, both the expected silaketal 16 and the corresponding diol 15 were obtained after chromatography (entry 3a). In later experiments, therefore, the crude cyclic silaketals were treated with HF·pyridine before purification. Using this protocol, the silaketals 7f, 7h and 7l yielded the diols 14, 15 and 18 (entries 2, 3b and 5).
For the substrates 7d, 7f, 7h and 7l, the initial metathesis product was a seven- to nine-membered cyclic silaketal. Similar ring-closing metatheses are well known.2–4 However, in the case of the silaketal 7j, the outcome of the metathesis reaction was remarkable: the diol 17 was isolated as a 55 : 45 E : Z mixture (entry 4). The fate of 7j is consistent with the macrocycle-closing metathesis reaction (→ 20) which is described in Scheme 4. Cyclisation of 19 to form a six-membered ring was not observed: presumably six-membered ring formation is reversible, and macrocyclisation (19 → 20) allows more efficient release from the fluorous tag.
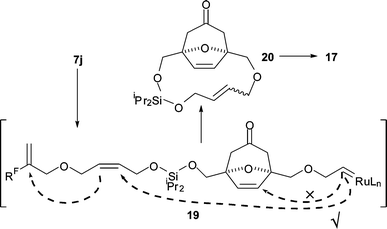 | ||
| Scheme 4 Macrocycle-closing metathesis of the silaketal 7j. | ||
In summary, we have developed an efficient method for the formation of unsymmetrical silaketals, which we applied in the synthesis of the fluorous-tagged metathesis substrates 7a–l. The method is remarkably chemoselective, and can be used to prepare silaketals with alkenyl and electron-rich aromatic substituents. We also observed a macrocycle-closing metathesis reaction that proceeded via a 13-membered cyclic silaketal. Through the use of a temporary silicon connection, therefore, it was possible to effect the net cross-metathesis of an unsaturated alcohol in which the reacting alkene was rather remote from the tether. Similar macrocycle-closing metathesis reactions may be useful in the synthesis of other compounds in which an alkene is remote from a potential tethering point.
Acknowledgements
We thank the Wellcome Trust, EPSRC, the University of Leeds and GSK for funding, James Titchmarsh for HPLC analyses, Simon Barrett for NMR analysis, and Dr Gordon McKiernan for helpful discussions.Notes and references
- M. Bols and T. Skrydstrup, Chem. Rev., 1995, 95, 1253 CrossRef CAS.
- (a) P. van de Weghe, D. Aoun, J.-G. Boiteau and J. Eustache, Org. Lett., 2002, 4, 4105 CrossRef CAS; (b) P. A. Evans, J. Cui, S. J. Gharpure, A. Polosukhin and H.-R. Zhang, J. Am. Chem. Soc., 2003, 125, 14702 CrossRef CAS; (c) T. Gaich and J. Mulzer, Org. Lett., 2005, 7, 1311 CrossRef CAS.
- B. A. Harrison and G. L. Verdine, Org. Lett., 2001, 3, 2157 CrossRef CAS.
- P. A. Evans, J. Cui and G. P. Buffone, Angew. Chem., Int. Ed., 2003, 42, 1734 CrossRef CAS.
- (a) C. Colombier, T. Skrydstrup and J. M. Beau, Tetrahedron Lett., 1994, 35, 8167 CrossRef CAS; (b) G. Stork and P. F. Keitz, Tetrahedron Lett., 1989, 30, 6981 CrossRef CAS; (c) J. W. Gillard, R. Fortin, H. E. Morton, C. Yoakim, C. A. Quesnelle, S. Daignault and Y. Guindon, J. Org. Chem., 1988, 53, 2602 CrossRef CAS.
- S. G. Leach, C. J. Cordier, D. Morton, G. J. McKiernan, S. Warriner and A. Nelson, J. Org. Chem., 2008, 73 DOI:10.1021/jo7026273.
- W. Zhang and D. P. Curran, Tetrahedron, 2006, 62, 11837 CrossRef CAS.
- J.-D. Moriggi, L. J. Brown, J. L. Castro and R. C. D. Brown, Org. Biomol. Chem., 2004, 2, 835 RSC.
- M. Petit, G. Chouraqui, C. Aubert and M. Malacria, Org. Lett., 2003, 5, 2037 CrossRef CAS.
- H. D. Maynard and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 4137 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and copies of NMR spectra for key compounds. See DOI: 10.1039/b804769n |
| This journal is © The Royal Society of Chemistry 2008 |