Hot off the press
Hot off the Press highlights recently published work for the benefit of our readers. Our contributors this month have focused on protein–protein interaction networks, the development of specific kinase inhibitors and the identification of biomarkers for the diagnosis of Alzheimer's disease. New contributors are always welcome. If you are interested please contact molbiosyst@rsc.org for more information, we'd like to hear from you.
Networking with drugs
Reviewed by: Arthur Wuster, MRC Laboratory of Molecular Biology, Cambridge, UK. E-mail: awuster@mrc-lmb.cam.ac.uk
Biological networks such as protein–protein interaction networks have become valuable tools for drug discovery. According to the database DrugBank, there are now around 1200 drugs approved by the United States Food and Drug Administration (FDA), with another 3100 drugs under investigation. Together the approved drugs target around 400 human proteins.
Recently Yıldırım et al. from Marc Vidal's and Albert-László Barabási's group represented the interactions between these target proteins and the drugs in a bipartite drug-target network. From this, they derived two more networks called the target protein network and the drug network (Fig. 1).
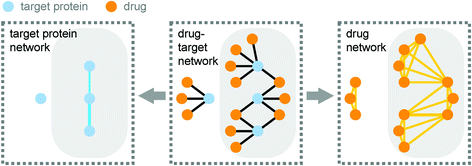 | ||
| Fig. 1 A bipartite network of drugs and their target proteins (drug-target network; centre panel) contains the information to derive both the target protein network and the drug network. In the target protein network (left panel) two proteins are linked if they are targeted by the same small molecule. In the drug network (right panel) two drugs are linked if they target the same protein. The giant component of each network is highlighted in grey. Reprinted by permission from Macmillan Publishers Ltd: Nat. Biotechnol., 2007, 25, 1119–1126, copyright 2007. | ||
Yıldırım et al. showed that it is possible to extract meaningful information about drugs and the drug discovery process from these networks. Because many drugs have more than one target protein, and since many proteins are targeted by more than one drug, many of the nodes of these networks are connected in a single component. However, this giant component of both the target protein network and the drug network is smaller than what would be expected by chance. This is because nodes with a lot of connections to other nodes are preferentially linked to each other, which indicates that drugs often target proteins which have previously been targeted by other drugs. In contrast, a target protein network derived from the drugs which are still under investigation by the FDA form a giant component which is larger than expected by chance. This might indicate that more recent drugs tend to target proteins which have not previously been targeted.
By integrating their data on drug-target interactions with genetic disease associations, gene expression data, and protein–protein interaction networks, the authors made interesting observations on the activity of drugs. For example, on the human protein-protein interaction network, a drug more often targets proteins which are either encoded by the disease gene or interact with the disease gene than would be expected by chance. When considering drugs which were approved after 1996, this trend is even more pronounced.
Altogether, the results suggest that there is a trend for more rational drug design over the last decade. However, this has not yet translated into an increased rate of FDA drug approval: Until now, the year with the most new approved drugs was 1996. Therefore, although drug design has become more rational it has not necessarily become more productive.
M. A. Yıldırım, K. I. Goh, M. E. Cusick, A. Barabási and M. Vidal, Nat. Biotechnol., 2007, 25, 1119–1126
Noncovalent tethering of a warhead to a phage-displayed library facilitates the identification of a highly selective inhibitor
Reviewed by: Hyun-Suk Lim, UT-Southwestern Medical Center, Dallas, Texas, USA.
Protein kinases play important roles in regulating most cellular functions and the deregulation of kinase activity is implicated in a number of diseases. Pharmacological inhibitors of protein kinase are of great interest and considerable efforts have been made to develop specific kinase inhibitors, most of which target the ATP-binding pocket of kinases. Although several potent and selective ATP-competitive inhibitors have been successfully developed and some of them are now clinically used, ATP-directed kinase inhibitors have several drawbacks. For example, since the ATP binding pocket is highly conserved across the protein kinase family, it would be difficult to identify specific kinase inhibitors that can discriminate among all the protein kinases as well as many other proteins that utilize ATP as a cofactor.
One possible approach towards highly specific kinase inhibitors would be the development of bisubstrate inhibitors by utilizing sites on protein kinases other than the ATP-binding pocket, for example, substrate-binding sites. The feasibility of the concept has been demonstrated previously by conjugating a ATP mimic and a known peptide substrate. For many kinases, however, there is no structural or peptide substrate information. The authors designed an in vitro bivalent selection approach by combining a promiscuous warhead with a phage-display library. This biological selection strategy will provide selective bivalent inhibitors of protein kinases, of which structural or peptide substrate information is unknown.
The group employed two interacting proteins, Jun and Fos, for a noncovalent tethering strategy to combine a warhead with a library. They conjugated Jun with a derivative of staurosporine as a kinase targeting warhead, which is a well-known ATP-competitive kinase inhibitor without selectivity. A phage-displayed peptide library was constructed adjacent to the Fos domain, which heterodimerizes with Jun. In each round of phage display selection, staurosporin derivative-Jun conjugate was mixed with a Fos-conjugated cyclic peptide library and exposed to the immobilized c-AMP-dependent protein kinase (PKA). They identified a cyclic peptide from six round of selection. This cyclic peptide was then covalently coupled to the staurosporin derivatives through a polyethylene glycol (PEG) linker to provide a bivalent molecule (Fig. 2). Based on PKA inhibition assay, the bivalent molecule showed considerably increased inhibitory activity with an IC50 of 2.6 nM, compared to its parent compounds: staurosporin derivative (IC50 = 243 nM) and the cyclic peptide (IC50 = 57 μM). These results indicate that the resulting bivalent compound binds to the PKA cooperatively with a synergistic effect as expected. Furthermore, this compound showed high specificity to PKA while the warhead itself, a staurosporine derivative, did not discriminate among five distinct kinases.
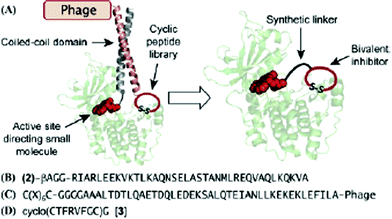 | ||
| Fig. 2 Bivalent inhibitor selection strategy: (A) Noncovalent tethering of staurosporine with a phage-display peptide library through a coiled-coil heterodimer for targeting a kinase, PKA. The selected peptide can be subsequently conjugated to staurosporine to provide a bivalent kinase inhibitor. (B) Staurosporine derivative 2 conjugated to the Jun. (C) Phagedisplayed cyclic peptide library attached to Fos (X) any of the 20 natural amino acids). (D) Selected cyclic peptide 3. Reprinted with permission from J. Am. Chem. Soc., 2007, 129, 13812. Copyright 2007 American Chemical Society. | ||
This study reports an elegant example of utilizing “a fragment-based bivalent ligand selection methodology.” However, peptide-based libraries have several limitations for targeting intracellular proteins due to their poor cell permeability and cellular stability. Nonetheless, this methodology would be a useful and general tool to identify high-affinity protein kinase inhibitors since the isolated inhibitors highly specific to any given proteins can be useful as probes in proteomics such as protein-capture microarrays.
Meyer, S. C., Shomin, C. D., Gaj, T. and Ghosh, I. Tethering small molecules to a phage display library: Discovery of a selective bivalent inhibitor of protein kinase A. J. Am. Chem. Soc., 2007, 129, 13812–13813.
Biomarkers for the early diagnosis of Alzheimer's disease
Reviewed by: John Astle, UT-Southwestern Medical Center, Dallas, Texas, USA
Alzheimer's disease is the most common form of dementia, a disease that affects millions over the age of 65. Despite its prevalence, diagnosis of Alzheimer's disease can sometimes be more of an art than a science, with exclusion of other diseases playing the major role in the diagnostic process. The only way to definitively confirm the diagnosis of Alzheimer's disease is an autopsy. New clinical tests that would aid in diagnosis of this debilitating disease, particularly at early stages of the disease, are needed. A recent discovery by Ray et al. of a panel of serum biomarkers may provide such a test.
Many diseases are accompanied by a release of specific proteins or metabolites into the blood, and the authors of this study began their biomarker hunt with the hypothesis that this would hold true for Alzheimer's disease. They used serum samples from 43 Alzheimer's patients and 40 “non-demented” controls as a training set to screen a panel of 120 signaling proteins using arrayed sandwich ELISAs. They identified 19 proteins with either increased or decreased expression in the disease vs. control groups. The significance of these 19 was determined by a procedure known as the significance analysis of microarrays (SAM). 18 of the 19 were independently identified using a separate algorithm called predictive analysis of microarrays (PAM), and these 18 had 95% positive agreement and 83% negative agreement with the clinical diagnoses.
To validate these biomarkers, PAM analysis was performed on an independent test set containing 42 Alzheimer's, 39 non-demented, and 11 demented but non-Alzheimer's serum samples. These samples were classified with 90% positive agreement and 88% negative agreement with clinical diagnoses, and 10 of the 11 demented but non-Alzheimer's samples were classified as non-Alzheimer's.
Perhaps the most important finding was that the expression profile of these 18 proteins in patients with mild cognitive impairment (MCI), but without clinical criteria sufficient for a diagnosis of Alzheimer's disease, was able to predict fairly accurately which patients would progress to Alzheimer's. 20 of 22 MCI patients who eventually progressed to Alzheimer's disease were classified correctly, and all 8 patients who progressed to other dementias were classified as non-Alzheimer's. 17 patients maintained MCI status at the time this study was published, and it will be interesting to see how well the biomarkers predict eventual progression to clinical Alzheimer's disease in these patients.
If these findings hold true in larger, independent patient populations, physicians will have a wonderful new tool to help diagnose patients at earlier stages, which would help prepare patients and their families, as well as decrease the financial burden on society by beginning treatment early and delaying the necessity for costly assisted living programs. Further investigation into the role of these 18 signaling proteins in the disease process may also lead to new treatments for this disease.
S. Ray, M. Britschgi, C. Herbert, Y. Takeda-Uchimura, A. Boxer, K. Blennow, L. F. Friedman, D. R. Galasko, M. Jutel, Anna Karydas, J. A. Kaye, J. Leszek, B. L. Miller, L. Minthon, J. F. Quinn, G. D. Rabinovici, W. H. Robinson, M. N. Sabbagh, Y. T. So, D. L. Sparks, M. Tabaton, J. Tinklenberg, J. A. Yesavage, R. Tibshirani, T. Wyss-Coray. Nat Med., 2007, 13(11), 1359–62.
Hot off the RSC press
Catalysis in a cavity
Reviewed by: Nicola Burton, Royal Society of Chemistry, Cambridge, UK.
Chemists in the US have created a molecular vase that mimics an enzyme's catalytic activity.
Chemical catalysts are chemoselective—they are able to recognise and transform a particular functional group—but few are capable of differentiating differently sized or shaped molecules with the same functional groups. This is where enzymes have an advantage. They are usually very specific about which reactions they catalyse: the shape of the starting material and its interaction with the enzyme being important factors in that specificity.
Julius Rebek, Jr. and Richard Hooley from the Scripps Research Institute, La Jolla, California, have set themselves the goal of creating chemical systems that match the selective catalytic abilities of enzymes whilst retaining the properties of normal chemical catalysts.
The researchers synthesised a vase-shaped molecule, called a cavitand, and used it to catalyse a Diels–Alder reaction between an unsaturated imide and an aromatic alcohol.
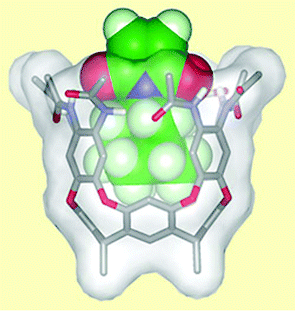 | ||
| Fig. 3 An imide molecule fits snugly in the molecular cavity perfectly primed to react with an alcohol. | ||
Rebek's system mimics an enzyme in that the cavitand has a cavity in which only an appropriately sized starting material can fit. A hydrogen bonding network at the rim of the cavity, similar to that found in enzymes, activates the starting material—the unsaturated imide—by sucking electron density from the double bond. This increases its reactivity and accelerates its Diels–Alder reaction with the aromatic alcohol. The product is too big to fit in the cavity and is ejected, leaving the cavity free to activate another molecule of starting material.
Rebek aims to improve the cavitand so that its hydrogen bonding network is positioned directly at the bound starting material. This would allow more challenging reactions to be accelerated and catalysed.
Richard J. Hooley and Julius Rebek Jr., Org. Biomol. Chem., 2007, 5, 3631
DOI: 10.1039/b713104f
Substrate screening made simple
Reviewed by: Danièle Gibney, Royal Society of Chemistry, Cambridge.
A simple assay for identifying protease substrates will make such experiments accessible to everyone, say scientists in Switzerland.
Proteases are enzymes that hydrolyse peptide bonds, and are involved in many important processes in the body. Identifying protease substrates is important for studying the structure and function of these enzymes, and can also produce leads for drug development, explained Jean-Louis Reymond, a bioorganic chemist from the University of Bern. Proteases are implicated in diseases such as cancer, and viral proteases are also important drug targets, he said.
There are currently many techniques for screening protease substrates, but according to Reymond they all involve complex experiments and remain inaccessible to most laboratories. Following calls for a simpler method, he and colleague Jacob Kofoed have developed a system that Reymond says is 'very simple, very reliable, and can be repeated by students with a minimum of training.
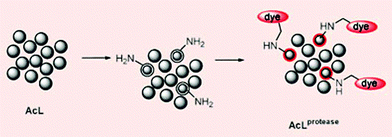 | ||
| Fig. 4 Members of a peptide library (AcL) are hydrolysed by a protease leaving free amino groups to react with a selective dye. | ||
In the new method a library of peptides carried on polymer beads is exposed to a protease. This step leaves a free amino group on the peptides that are hydrolysed by the enzyme. A selective dye then reacts with the amino groups, colouring the beads carrying protease substrates red. The peptide sequences on the red beads are then analysed, thus identifying the substrates.
Reymond and Kofoed used their method to find substrates for various proteases among a library of over 65 000 peptides. The results from the assay agreed with those found by other methods.
Reymond said he would be able to use the technique in several projects, including studying the function of the intestinal proteases meprin alpha and beta. To extend the method's potential uses he also plans to include non-natural peptides in the substrate libraries. ‘This should provide peptides with higher selectivities for their target,’ said Reymond.
Jacob Kofoed and Jean-Louis Reymond, Chem. Commun., 2007, 4453
DOI: 10.1039/b713595e
| This journal is © The Royal Society of Chemistry 2008 |