DOI:
10.1039/B713310C
(Paper)
J. Mater. Chem., 2008,
18, 91-97
Thick wall mesoporous carbons with a large pore structure templated from a weakly hydrophobic PEO–PMMA diblock copolymer†
Received
30th August 2007
, Accepted 28th September 2007
First published on 22nd October 2007
Abstract
Thick wall mesoporous carbons (FDU-18) with ordered large pore structures were synthesized via an evaporation induced self-assembly (EISA) approach by using a novel amphiphilic diblock copolymer with a weakly hydrophobic segment as a template, soluble resol as a carbon source and THF as a solvent. The versatile atom transfer radical polymerization (ATRP) method was utilized to prepare the diblock copolymer, poly(ethylene oxide)-b-poly(methyl methacrylate) (PEO-b-PMMA). 1H NMR spectroscopy and gel permeation chromatography show that the obtained PEO-b-PMMA has a Mn of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 070 g mol−1 and a narrow molecular weight distribution (1.09). Small angle X-ray scattering (SAXS) and transmission electron microscopy (TEM) measurements demonstrate that the obtained mesoporous carbons have a face centered cubic close-packed mesostructure (Fm
070 g mol−1 and a narrow molecular weight distribution (1.09). Small angle X-ray scattering (SAXS) and transmission electron microscopy (TEM) measurements demonstrate that the obtained mesoporous carbons have a face centered cubic close-packed mesostructure (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) with a large unit cell (∼38 nm). N2 sorption isotherms indicate that the mesoporous carbons have a high surface area up to 1050 m2 g−1 and a uniform large mesopore (∼10.5 nm). It is interesting that the pore walls of the mesoporous carbonaceous materials are ultra thick (ca. 11.5–12.4 nm), which are even larger that their pore diameters. The thick pore walls are related to the weakly hydrophobic block PMMA, which can associate with the phenolic resol precursors. Many useful performances such as high mechanical/chemical stability and graphitization could be expected for this kind of thick wall carbon material.
m) with a large unit cell (∼38 nm). N2 sorption isotherms indicate that the mesoporous carbons have a high surface area up to 1050 m2 g−1 and a uniform large mesopore (∼10.5 nm). It is interesting that the pore walls of the mesoporous carbonaceous materials are ultra thick (ca. 11.5–12.4 nm), which are even larger that their pore diameters. The thick pore walls are related to the weakly hydrophobic block PMMA, which can associate with the phenolic resol precursors. Many useful performances such as high mechanical/chemical stability and graphitization could be expected for this kind of thick wall carbon material.
Introduction
In the past few years, ordered mesoporous carbons, owing to their high surface area, large pore volume, and high thermal, chemical and mechanical stability, have received considerable attention for their high potential applications in separation, catalysis, gas storage, electrode materials and so on.1–7 Considering the application requirements, carbons with large mesopores and thick walls are highly desirable because they hold great promise in fields involving the transport of large molecules, but also provide opportunity for further functionalization and modification such as to produce graphitized carbon frameworks, to incorporate metal or metal oxides in the thick pore walls. Furthermore, the thick pore walls may also contribute high mechanical and chemical stability. To date, although great progress has been made in the synthesis of ordered mesoporous carbons by using conventional hard-templating approaches8–17 or the recently-developed organic–organic assembly (soft-templating) approaches,18–22 few cases succeed in tuning the pore sizes and pore wall thicknesses, and the synthesis of mesoporous carbons with large pores and thick walls remains particularly challenging. In the hard-templating cases, there is little chance to synthesize ordered mesoporous carbon replicas with adjustable pore size larger than 5 nm due to the difficulties in varying the pore wall thickness of the silica templates.17 In the case of organic–organic assembly approaches, Pluronic triblock copolymers PEO–PPO–PEO [PPO = poly(propylene oxide)] such as P123 and F127 were frequently used as structure-directing agents. However, the pore sizes and wall thicknesses of mesoporous carbons are limited to 4.0 and 7.0 nm, respectively, owing to the limitation of the molecular weight of the block copolymers.18,19
Since the pore sizes, wall thicknesses and pore symmetries of mesoporous materials are predominantly determined by parameters of the copolymer templates, such as total molecular weight, hydrophilicity/hydrophobicity ratio and volume fractions of each of the blocks, the design and synthesis of new block copolymers are highly desirable. In this regard, Dai and coworkers23 prepared highly ordered mesoporous carbons with 34 nm pore size and 10 nm wall thickness using the high molecular weight diblock copolymer polystyrene-block-poly(4-vinylpyridine) (PS-b-P4VP) and phenolic resol as a carbon source through a stepwise assembly approach. Very recently, we have prepared the amphiphilic diblock copolymer poly(ethylene oxide)-block-polystyrene (PEO125-b-PS230) via atom transfer radical polymerization (ATRP). By using the PEO-b-PS as a template, we successfully synthesized highly ordered mesoporous carbon (FDU-18) with a pore size of 23 nm and wall thickness of ∼9.9 nm, through solvent evaporation induced assembly approach (EISA).24 However, the carbon walls are thin (thickness <10 nm). Up to now, almost all of the reported mesoporous materials obtained either through the surfactant-templating approach or by using mesoporous silica as a hard-template have pore sizes larger than their respective wall thickness.
In this paper, we report the synthesis of highly ordered mesoporous carbon (FDU-18) by using a weakly hydrophobic diblock copolymer, poly(ethylene oxide)-b-poly(methyl methacrylate) (PEO-b-PMMA) as a template, soluble resol as a carbon source and THF as a solvent, via an EISA method. ATRP technique was utilized to prepare the PEO-b-PMMA diblock copolymer. Small angle X-ray scattering (SAXS) and transmission electron microscopy (TEM) measurements show that the obtained mesoporous carbon has a face centered cubic close-packed mesostructure (Fmm). N2 sorption isotherms indicate that the mesoporous carbon have a high surface area up to 1050 m2 g−1, a uniform large pore (∼10.5 nm). Interestingly, the obtained FDU-18 have very thick pore walls (11.5–12.4 nm), which is even larger than the pore size. The ultra thick wall may not only offer mesoporous carbons with high chemical/mechanical stability which is desirable in practical applications, but also favors the incorporation of metal (oxide) particles in the carbon framework with unique electronic or magnetic properties.
Experimental section
Chemicals
Monomethoxy poly(ethylene oxide)s (Monomethoxy PEO5000, molecular weight = 5000) and 2-bromoisobutyryl bromide were phased from Aldrich. N,N,N′,N′,N″-pentamethyldiethylenetriamine (PMDETA) was purchased from Acros. Methyl methacrylate (MMA), tetrahydrofuran (THF) (>99%), pyridine (>99%), copper bromide (CuBr), and anhydrous ether (>99%) were purchased from Shanghai Chemical Corp. MMA was treated by filtering through an Al2O3 column to remove any polymerization inhibitors. Millipore water was used in all experiments.
Preparation of PEO-b-PMMA diblock copolymer.
The PEO-b-PMMA diblock copolymer was prepared using the ATRP technique. Firstly, macroinitiator PEO-Br was prepared as follows: 20.0 g of monomethoxy PEO5000 was dissolved in 100 mL of tetrahydrofuran (THF)–pyridine mixture (6 : 4 volume ratio). The obtained solution was cooled in an ice–water bath (0 °C), 3.00 g of 2-bromoisobutyryl bromide (13 mmol, Aldrich) was dropwise added to the solution under stirring for 30 min. The resultant solution was further stirred at 30 °C overnight. After cooling to room temperature, 200 mL of cold ether was added to the solution. The white product of PEO-Br was precipitated from the reaction solution, washed with cold ether and further dried under vacuum. Then, PEO-b-PMMA diblock copolymer was prepared viaATRP by using PEO-Br as an initiator as follows: 3.00 g of PEO-Br (0.53 mmol), 0.08 g of CuBr (0.57 mmol), 0.10 g of PMDETA (0.57 mmol) and 20.0 g of methyl methacylate (200 mmol) were added to an ampoules bottle. The bottle containing reactants was fully degassed with three freeze–pump–thaw cycles and sealed under vacuum. It was subsequently immersed in a thermostated oil bath at 60 °C under stirring to allow polymerization of MMA. After reaction for 1 h, the system was cooled down to room temperature. The gel-like product was dissolved in 50 mL of THF and filtered through an Al2O3 column to remove the Cu complex. 200 mL of cold ether was poured into the solution to precipitate PEO-b-PMMA diblock copolymer. The copolymer product was then dried in vacuum. The yield was ∼38 wt% based on the feeding MMA amount.
Synthesis of mesoporous carbon FDU-18.
Mesoporous carbons FDU-18 were prepared through an EISA strategy by using the obtained PEO-b-PMMA diblock copolymer as a template, THF as a solvent and resol as a carbon source. The resol precursor soluble phenolic resin, with low molecular weight (Mn < 500) was prepared from phenol and formaldehyde in a base-catalyzed process according to a procedure reported previously.18 0.61 g of phenol was melted at 40–42 °C in a flask and mixed with 0.13 g of 20 wt% sodium hydroxide (NaOH) solution under stirring. After 10 min, 1.05 g formalin (37 wt% formaldehyde) was added below 50 °C. Upon further stirring for 1 h at 70–75 °C, the pH was adjusted with 0.6 M HCl solution until it reached a value of ∼7.0, and water was removed by vacuum evaporation below 50 °C. The final product was dissolved in ethanol. The molar ratio of phenol : formaldehyde : NaOH was 1 : 2 : 0.1.
Ordered mesoporous carbon FDU-18 could be synthesized in the range of phenol : formaldehyde : PEO-b-PMMA (molar ratio) = 1 : 2 : 0.00176–0.00392. For a typical synthesis, 1.0 g (containing 1.33 mmol phenol and 2.66 mmol formaldehyde) of the resol–THF solution (20 wt%) was added to 2.4 g (0.0024 mmol) of PEO-b-PMMA/THF solution (2.0 wt%) with stirring to form a homogeneous solution. Transparent membranes were obtained by pouring the solution into Petri dishes to evaporate THF at room temperature for 12 h, followed by further heating in an oven at 100 °C for 24 h. The as-made product was scraped and crushed into a powder. Pyrolysis was carried out in a tubular furnace under N2 at different temperatures (450–1000 °C) for 3 h. The heating rate was 1 °C min−1 below 450 °C and 5 °C min−1 above 600 °C. The final products were designated as FDU-18-X, where X refers to the calcination temperature.
Characterization.
1H-NMR spectra were recorded at 25 °C on a DMX 500 MHz spectrometer (Bruker, Germany) with tetramethylsilane as an internal standard and CDCl3 as a solvent. Gel permeation chromatography (GPC) was performed on an Agilent 1100 gel permeation chromatographer with a refractive index detector and UV-vis detector (wavelength 190–950 nm, USA) with the use of THF as an eluent (1.0 mL min−1). GPC was calibrated with a monodisperse polystyrene standard. Fourier transform infrared (FT-IR) spectra were collected on Nicolet Fourier spectrophotometer using KBr pellets (USA). Thermogravimetric (TG) analysis was carried out on a Mettler Toledo TGA-SDTA851 analyzer (Switzerland) from 25 to 900 °C under nitrogen with a heating rate of 5 °C min−1. Small angle X-ray scattering (SAXS) measurements were taken on a Nanostar U small angle X-ray scattering system (Bruker, Germany) using Cu Kα radiation (40 kV, 35 mA). The d-spacing values were calculated by the formula d = 2π/q, where q is the scattering vector. The wall thickness was calculated from WT = √2a/2 −D, where a represents the cell parameter and D is the pore diameter calculated from the N2 sorption measurements. Nitrogen sorption isotherms were measured at 77 K with a Micromeritics Tristar 3000 analyzer (USA). Before measurements, the samples were degassed in a vacuum at 200 °C for at least 6 h. The Brunauer–Emmett–Teller (BET) method was utilized to calculate the specific surface areas. By using the Broekoff-de Boer (BdB) model, the pore volumes and pore size distributions were derived from the adsorption branches of isotherms, and the total pore volumes (Vt) were estimated from the adsorbed amount at a relative pressure P/P0 of 0.992. Transmission electron microscopy (TEM) experiments were conduced on a JEOL 2011 microscope (Japan) operated at 200 kV. Raman spectra were obtained with a Dilor LabRam-1B microscopic Raman spectrometer (France), using a He–Ne laser with an excitation wavelength of 632.8 nm. The C, H, and O contents were measured on a Vario EL III elemental analyzer (Germany).
Results and discussion
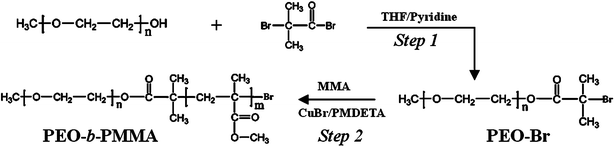
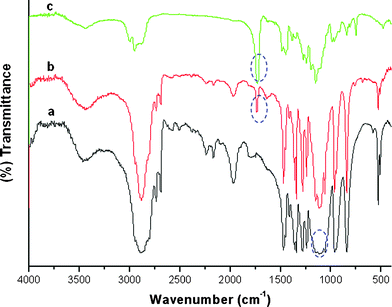
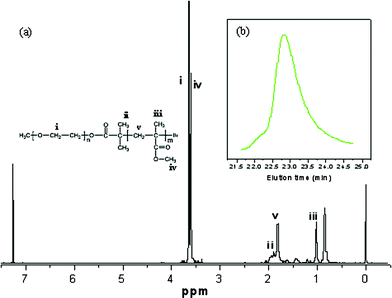
The diblock copolymer PEO-b-PMMA is easily prepared by using a versatile ATRP technique which is readily accessible for those who do not have any polymer synthesis experience. The synthesis procedure (Fig. 1) is similar to that for PEO-b-PS, including the preparation of the macroinitiator PEO-Br by reacting monomethoxy PEO5000 with 2-bromoisobutyryl bromide (Step 1), and the polymerization of methyl methacrylate (MMA) (Step 2). The FT-IR spectra display absorption bands at 1109 cm−1 originating from the EO units of the PEO precursor, a carbonyl stretching band at 1735 cm−1 for the macroinitiator PEO-Br formed by the acetylation reaction, and a strong carbonyl stretching band at 1735 cm−1 assigned to the PMMA segment (Fig. 2). These characteristic bands demonstrated a successful preparation of the macroinitiator PEO-Br and the diblock copolymer PEO-b-PMMAvia the ATRP method. The chemical structure and composition of the obtained diblock copolymer PEO-b-PMMA were determined from the 1H NMR spectrum (Fig. 3a). The signals around 3.60 (i) and 1.90 ppm (ii) are associated with EO units and the –CH3 which originated from PEO5000 and 2-bromoisobutyryl bromide, respectively. The signals at 0.8–1.2, 1.7–2.0, 3.6 ppm are assigned to the repeated MMA units. The GPC analysis indicates that the obtained diblock copolymer has a polydispersity index (PDI) of 1.09, suggesting a monodisperse molecular weight distribution (Fig. 3b). The Mn value of the block copolymers is calculated to be 20070 g mol−1 according to the 1H NMR results. Therefore the composition of the diblock copolymer can be approximately formulated as PEO125-b-PMMA144. The TG analysis (see Fig S1a, ESI†) indicates that the PEO125-b-PMMA144copolymer can be decomposed at ca. 340–420 °C under N2. A minor residue (about 4.3 wt%) is observed, suggesting that the diblock copolymer used as a template can be easily removed by pyrolysis.
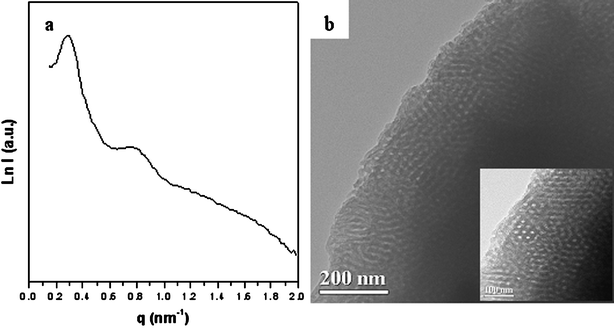
The as-made FDU-18 shows a poor SAXS pattern (Fig. 4a), probably due to the low contrast between the PEO-b-PMMA templates and the phenolic resin frameworks. After pyrolysis at 450 °C in N2, the resulting FDU-18-450 displays a well-resolved SAXS pattern with four scattering peaks at q-values of 0.29, 0.48, 0.78, 1.34 nm−1 (Fig. 4b). These scattering peaks can be indexed to the 111, 220, 422 and 800 reflections of a face centered cubic (fcc) mesostructure with the space group of Fmm, indicating a highly ordered mesostructure. The cell parameter (a) is calculated to be 37.5 nm (Table 1), suggesting a large unit cell of the mesostructure. A similarly well-resolved SAXS pattern is observed for FDU-18-600 after pyrolysis at 600 °C (Fig. 4c), but it causes a structural shrinkage of about 9.3% as calculated from the unit cell values (Table 1). After pyrolysis at 800 °C, the well-resolved SAXS pattern is retained (Fig. 4d). Even after further pyrolysis at a higher temperature of 1000 °C, a resolved SAXS pattern is still observed, indicating that the mesoporous carbon FDU-18 is thermally stable (Fig. 4e). Compared to FDU-18-450, mesoporous carbon FDU-18-800 has a smaller cell parameter of 32.4 nm, reflecting a 13% shrinkage in the framework caused by the carbonization of the phenolic resin framework. After pyrolysis at 1000 °C, the cell parameter of FDU-18-1000 further decreases to 30.0 nm, revealing further framework shrinkage of about 7%. TEM images and corresponding Fourier diffractograms of mesoporous carbon FDU-18-800 show a high degree of periodicity over large domains, viewed from the [100], [110] and [211] directions (Fig. 5), further confirming that the carbon mesostructure templated by the diblock copolymer PEO-b-PMMA possesses ordered cubic (fcc) symmetry. As clearly shown in these images, the obtained mesoporous carbon has uniform and large spherical mesopores. It suggests that the diblock copolymers assemble into globular micelles and undergo spherical packing during the EISA process. The cell parameter of the FDU-18-800 estimated from the TEM images is about 32.0 nm, agreeing well with that from the SAXS pattern. In addition, as shown in the TEM images, especially for the images viewed from [100] and [110] (Fig. 5a, b), the obtained mesoporous carbon has a very thick pore wall of about 12.0 nm, as roughly determined from the images.
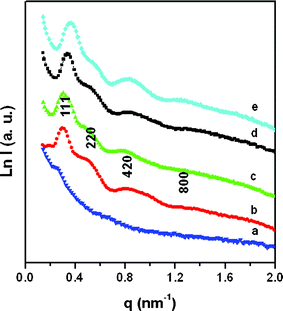 |
| | Fig. 4
SAXS patterns of (a) as-made mesoporous carbon FDU-18, (b) FDU-18-450, (c) FDU-18-600, (d) FDU-18-800 and (e) FDU-18-1000. | |
![TEM images of mesoporous carbon FDU-18-800 viewed from the [100] (a, b), [110] (c) and (d) [211] directions with respective diffractograms.](/image/article/2008/JM/b713310c/b713310c-f5.gif) |
| | Fig. 5
TEM images of mesoporous carbon FDU-18-800 viewed from the [100] (a, b), [110] (c) and (d) [211] directions with respective diffractograms. | |
Table 1 Textural properties of mesoporous carbon FDU-18 templated by PEO125-b-PMMA144diblock copolymera
| |
d
111
/nm |
a/nm |
Pore diameter/nm |
Wall thickness/nm |
BET surface area/m2 g−1 |
Micropore surface/m2 g−1 |
Total pore volume/cm3 g−1 |
Micropore volume/cm3 g−1 |
|
The d-spacing (d111) values were calculated by the formula d = 2π/q, and the unit cell parameters (a) were calculated from the formula a = d111√3. The wall thickness values were calculated from WT = √2a/2 −D, where a is the cell parameter and D is the mesopore diameter.
|
|
FDU-18-450
|
21.0 |
37.5 |
12.8 |
13.7 |
570 |
399 |
0.367 |
0.177 |
|
FDU-18-600
|
19.6 |
34.0 |
11.6 |
12.4 |
698 |
535 |
0.392 |
0.195 |
| FDU-18-800 |
18.7 |
32.4 |
10.5 |
12.4 |
1050 |
856 |
0.57 |
0.38 |
|
FDU-18-1000
|
17.3 |
30.0 |
9.7 |
11.5 |
1580 |
1360 |
0. 82 |
0.62 |
N2 sorption isotherms of the mesoporous carbonaceous FDU-18-450 exhibit representative type IV curves with a sharp capillary condensation step in the relative pressure range 0.72–0.82 (Fig. 6Aa), indicative of a large and uniform mesopore. A large H2-type hysteresis loop with delayed capillary evaporation at a relative pressure of about 0.5 is observed, implying a caged mesopore with a window size smaller than 4.0 nm. According to the BdB sphere model, the mean pore diameter determined from the adsorption branch is 12.8 nm (Fig. 6Ba). The BET surface area and pore volume are 570 m2 g−1 and 0.37 cm3 g−1, respectively (Table 1). After pyrolysis at 600 °C in a N2 atmosphere, FDU-18-600 displays similar isotherms with a large H2-type hysteresis loop. The mean mesopore size calculated from the BdB sphere model is about 11.6 nm (Fig. 6Bb), smaller than that of FDU-18-450, which is due to framework shrinkage during pyrolysis. As the pyrolysis temperature further increases to 800–1000 °C, the pore size decreases to 9.7 nm. In contrast, the BET surface area and pore volume dramatically increase with the increase of the pyrolysis temperature (Table 1). FDU-18-1000 has high BET surface area up to 1580 m2 g−1 and pore volume (0.82 cm3 g−1), which are about two times higher than that for FUD-18-450. The t-plot calculation reveals that the micropore surface area of FDU-18-1000 is 1360 m2 g−1, three times larger than the value (399 m2 g−1) of FDU-18-450 (Table 1). These results imply that numerous micropores are generated during the high temperature pyrolysis. It is caused by dehydrogenation and deoxygenation from the polymeric frameworks during the pyrolysis. N2 sorption isotherms (Fig. 6A) also show that all of the FDU-18 samples have large H2-type hysteresis loops at a relative pressure (P/P0) of about 0.5 with delayed capillary evaporation, corresponding to caged mesopores with a window size smaller than 4.0 nm.
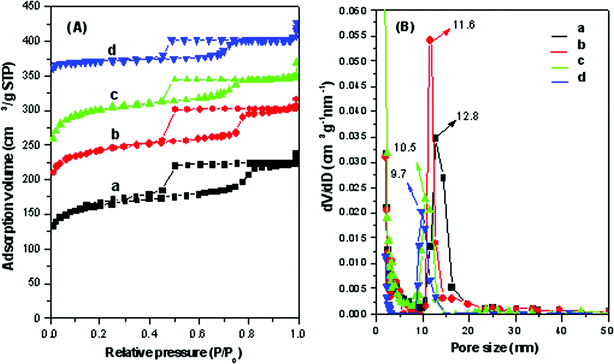 |
| | Fig. 6 (A) N2 adsorption–desorption isotherms and (B) pore size distribution curves of (a) FDU-18-450 pyrolyzed at 450 °C in N2, (b) FDU-18-600 pyrolyzed at 600 °C in N2, (c) FDU-18-800 pyrolyzed at 800 °C in N2 and (d) FDU-18-800 pyrolyzed at 1000 °C in N2. | |
Based on the N2 sorption and SAXS results, the pore wall thickness of mesoporous carbonaceous material FDU-18-450 is as large as 13.7 nm, which is much larger than that of mesoporous silicates reported previously. The value of the wall thickness is even slightly larger than its mesopore diameter (12.8 nm) (Table 1). As the pyrolysis temperature increases from 600 to 800 °C, the pore wall thickness decreases to 12.4 nm, still larger than the corresponding mesopore diameter (10.5 nm). Notably, even after pyrolysis at 1000 °C, the resultant FDU-18-1000 has a large wall thickness (11.5 nm) (Table 1). Compared to the previously reported mesoporous carbons C-FDU-18 with mesopores of 23 nm and a wall thickness of 9.9 nm templated by PEO125-b-PS230diblock copolymer produced via a similar EISA approach,24 the FDU-18 prepared from PEO125-b-PMMA144 has a larger pore wall thickness despite the identical hydrophilic PEO chain length. Considering the identical mesostructures for both the carbon materials, the thicker pore wall of FDU-18 obtained here is possibly attributed to the weaker hydrophobicity of the PMMA block compared with the PS chains. To the best of our knowledge, ordered mesoporous carbons with such a thick wall have rarely been reported before. The thick wall would be beneficial to the improvement of the mechanical/chemical stability and incorporation of functional nanoparticles, and, as a result, the application and functionalization of mesoporous carbons.
The TG curve of the as-made FDU-18 templated by a weakly hydrophobic diblock copolymer PEO125-b-PMMA144 shows two weight loss stages with corresponding maximum temperatures at 200 and 410 °C, respectively (Fig. S1b, ESI†). The first one ends at ∼300 °C with weight loss of 17%, owing to the removal of minor adsorbed water and the dehydration of numerous hydroxyl groups in the phenolic resin framework. The second one ends at 480 °C with the weight loss of about 14%, mainly due to the decomposition of the PEO-b-PMMA template. The carbonization yield after the pyrolysis at 800 °C based on the resol precursor is about 53 wt%. Elemental analysis reveals that the framework of FDU-18-450 is composed of C (79.3 wt%), H (4.5 wt%) and O (16.2 wt%) with C : H : O of 6.6 : 4.5 : 1 in molar ratio, agreeing with the composition of the phenolic resins. It suggests that the FDU-18-450 possesses a polymer bakelite framework. After pyrolysis at 800 °C, the obtained FDU-18-800 shows a molar ratio for C : H : O of 16.0 : 3.5 : 1, suggesting a carbon framework. The FT-IR spectrum of the as-made FDU-18 shows strong vibration bands at 3400, 1730 and 1100 cm−1 assigned to –OH groups of the phenolic resins, PMMA and PEO blocks of the template, respectively (Fig. 7a). After pyrolysis at 450 °C, the bands at 1730 and 1100 cm−1 disappear, further revealing the removal of the PEO-b-PMMA template (Fig. 7b). Almost no absorption band is detected in the FT-IR spectrum of FDU-18-800, indicating the complete carbonization of the phenolic resins (Fig. 7c). The Raman spectrum (Fig. 8) of FDU-18-800 shows a G band at 1591 cm−1 and a D band at 1313 cm−1 with an intensity ratio IG/ID of 0.59, indicating that the products are mainly composed of amorphous carbon.
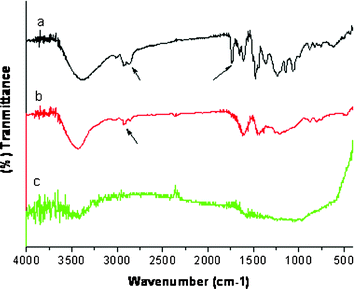 |
| | Fig. 7
FT-IR spectra of (a) the as-made mesoporous carbonaceous FDU-18, (b) FDU-18-450 obtained after pyrolysis at 450 °C and (c) FDU-18-800 obtained after pyrolysis at 800 °C. | |
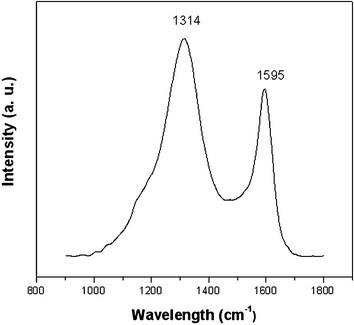 |
| | Fig. 8
Raman spectra of mesoporous carbon FDU-18-800 with a G band at 1595 cm−1 and a D band at 1314 cm−1 with an intensity ratio IG/ID of 0.59, suggesting the obtained mesoporous carbon has a low degree of graphitization. | |
On the basis of the above results, we speculate that the formation of the ordered mesoporous carbon FDU-18 is driven by hydrogen-bonding between the PEO125-b-PMMA144diblock copolymer and the resol molecules (Fig. 9). The resol precursors are low molecular weight (Mn <500) phenolic formaldehyde resins with a large amount of hydroxyl groups which can interact with the EO units of the diblock copolymer templates by hydrogen-bonds. After the thermal polymerization by heating at 100 °C, it can form three-dimensionally connected rigid frameworks. During the EISA process, PEO125-b-PMMA144diblock copolymers associated with resol molecules assemble into global micelles and pack into an fcc mesostructure.24 Due to the presence of ester groups in the PMMA segments, the resol molecules not only interact with the PEO chains but also partially associate with the PMMA segments to form the thick resin walls. By contrast, in the case of PEO125-b-PS230diblock copolymer, the PS segment forms an extremely hydrophobic core, and only the PEO chains interact with hydrophilic resol molecules. Therefore, despite the identical PEO chain length of the two diblock copolymer templates, more resol molecules can present around the hydrophobic phase in the PEO-b-PMMA/resol composites. Therefore, ordered mesoporous carbon frameworks with ultra thick walls can be obtained. Our results show that further increasing the molar ratio of resol : PEO-b-PMMA can result in poor or disordered carbonaceous mesostructures. The SAXS patterns and TEM images of the mesoporous carbonaceous sample prepared with a high phenol : formaldehyde : PEO-b-PMMA molar ratio of 1 : 2 : 0.00157 after pyrolysis at 450 °C show that the mesostructure is poorly regular and the short-range mesopore ordering is observed in small domains (Fig. 10). When an excess amount of resol precursor is used, numerous MMA units in the PEO125-b-PMMA144copolymer can interact with the hydrophilic resol molecules. Consequently, the intrinsic hydrophilic/hydrophobic balance of the PEO125-b-PMMA144copolymer is broken, and the copolymer composites are too hydrophilic to self-assemble into ordered mesostructures, leading to poor even disordered mesostructures during the EISA process.
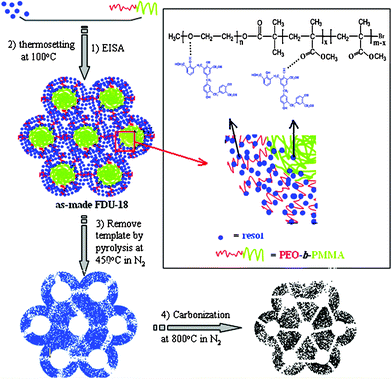 |
| | Fig. 9 Schematic illustration for the formation of the ordered mesoporous carbon FDU-18 with ultra thick pore walls. | |
Conclusions
A novel weakly hydrophobic diblock copolymer PEO-b-PMMA with monodisperse molecular weight distribution (Mn of 20070) was prepared by using a simple ATRP method. Using the obtained PEO125-b-PMMA144diblock copolymer as a template, ordered cubic mesoporous carbons (Fmm) with large cell parameters (32.4 nm) and high surface area (up to 1580 m2 g−1) have been synthesized through an EISA approach with phenolic resol as a carbon source. Because the weak hydrophobic PMMA block of the diblock copolymer template contains ester groups which can interact with hydrophilic phenolic resol precursors, the resultant mesoporous carbonaceous products have ultra thick pore walls (up to 13.7 nm). Interestingly the pore wall thickness is even larger than the pore diameter (∼10.5 nm). With the increase in the pyrolysis temperature, both the BET surface area and total pore volume increase, while the pore size and wall thickness decrease. The t-plots show that numerous micropores are generated during the pyrolysis. This weak hydrophobic templating process not only provides a way to tune the thickness, but also unique performances such as high mechanical/chemical stability are expected.
Acknowledgements
This work was supported by the NSF of China (20421303, 20641001 and 20521140450), the State Key Basic Research Program of the PRC (2006CB202502, 2006CB932302), the Shanghai Science & Technology Committee (06DJ14006, 055207078) and Shanghai Nanotech Promotion Center (0652nm024). YHD thanks the China Postdoctoral Scientific Fund and Shanghai Postdoctoral Scientific Program for financial support.
References
- F. Su, J. Zeng, X. Bao, Y. Yu, J. Y. Lee and X. S. Zhao, Chem. Mater., 2005, 17, 3960 CrossRef CAS.
- A. Lu, W. Schmidt, N. Matoussevitch, H. Bonnemann, B. Spliethoff, B. Tesche, E. Bill, W. Kiefer and F. Schüth, Angew. Chem., Int. Ed., 2004, 43, 4303 CrossRef CAS.
- T. Ohkubo, J. Miyawaki, K. Kaneko, R. Ryoo and N. A. Seaton, J. Phys. Chem. B, 2002, 106, 6523 CrossRef CAS.
- J. Lee, S. Yoon, T. Hyeon, S. M. Oh and K. B. Kim, Chem. Commun., 1999, 2177 RSC.
- S. Yoon, J. Lee, T. Hyeon and S. M. Oh, J. Electrochem. Soc., 2000, 147, 2507 CrossRef CAS.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169 CrossRef CAS.
-
J. W. Patrick, Porosity in Carbons: Characterization and Applications,, Edward Arnold, London, UK, 1995 Search PubMed.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743 CrossRef CAS.
- R. Ryoo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677 CrossRef CAS.
- A. H. Lu, W. Schmidt, A. Taguchi, B. Spliethoff, B. Tesche and F. Schüth, Angew. Chem., Int. Ed., 2002, 41, 3489 CrossRef CAS.
- M. Kaneda, T. Tsubakiyama, A. Carlsson, Y. Sakamoto, T. Ohsuna, O. Terasaki, S. H. Joo and R. Ryoo, J. Phys. Chem. B, 2002, 106, 1256 CrossRef CAS.
- C. H. Kim, D.-K. Lee and T. J. Pinnavaia, Langmuir, 2004, 20, 5157 CrossRef CAS.
- T. W. Kim, R. Ryoo, K. P. Gierszal, M. Jaroniec, L. A. Solovyov, Y. Sakamoto and O. Terasaki, J. Mater. Chem., 2005, 15, 1560 RSC.
- C. Z. Yu, J. Fan, B. Z. Tian, D. Y. Zhao and G. D. Stucky, Adv. Mater., 2002, 14, 1742 CrossRef CAS.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712 CrossRef CAS.
- T. W. Kim and L. A. Solovyov, J. Mater. Chem., 2006, 16, 1445 RSC.
- J. S. Lee, S. H. Joo and R. Ryoo, J. Am. Chem. Soc., 2002, 124, 1156 CrossRef CAS.
- Y. Meng, D. Gu, F. Q. Zhang, Y. F. Shi, H. F. Yang, Z. Li, C. Z. Yu, B. Tu and D. Y. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053 CrossRef CAS.
- Y. Meng, D. Gu, F. Q. Zhang, Y. F. Shi, L. Cheng, D. Feng, Z. X. Wu, Z. X. Chen, Y. Wan, A. Stein and D. Y. Zhao, Chem. Mater., 2006, 18, 4447 CrossRef CAS.
- F. Q. Zhang, Y. Meng, D. Gu, Y. Yan, C. Z. Yu, B. Tu and D. Y. Zhao, J. Am. Chem. Soc., 2005, 127, 13508 CrossRef CAS.
- F. Q. Zhang, Y. Meng, D. Gu, Y. Yan, C. Z. Yu, B. Tu and D. Y. Zhao, Chem. Mater., 2006, 18, 5279 CrossRef CAS.
- Y. Huang, H. Q. Cai, T. Yu, F. Q. Zhang, F. Zhang, Y. Meng, D. Gu, Y. Wan, X. L. Sun, B. Tu and D. Y. Zhao, Angew. Chem., Int. Ed., 2007, 46, 1089 CrossRef CAS.
- C. Liang, K. Hong, G. A. Guiochon, J. M. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785 CrossRef CAS.
- Y. H. Deng, T. Yu, Y. Wan, Y. F. Shi, Y. Meng, D. Gu, L. J. Zhang, Y. Huang, C. Liu, X. J. Wu and D. Y. Zhao, J. Am. Chem. Soc., 2007, 129, 1690 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2008 |
Click here to see how this site uses Cookies. View our privacy policy here.