NMR crystallography of p-tert-butylcalix[4]arene host–guest complexes using 1H complexation-induced chemical shifts
Darren H.
Brouwer
,
Saman
Alavi
and
John A.
Ripmeester
*
Steacie Institute for Molecular Sciences, National Research Council, 100 Sussex Drive, Ottawa ON, Canada K1A 0R6. E-mail: john.ripmeester@nrc-cnrc.gc.ca; Fax: +1 613 998 7833; Tel: +1 613 993 2011
First published on 16th May 2008
Abstract
1H magic-angle spinning (MAS) NMR spectra of p-tert-butylcalix[4]arene inclusion compounds with toluene and pyridine show large complexation-induced shifts of the guest proton resonances arising from additional magnetic shielding caused by the aromatic rings of the cavities of the host calixarene lattice. In combination with ab initio calculations, these observations can be employed for NMR crystallography of host–guest complexes, providing important spatial information about the location of the guest molecules in the host cavities.
The determination of structure in host–guest chemistry by diffraction methods is often challenging because of problems of disorder, which can be static or dynamic. Especially when such materials are designed for processes, understanding structure and dynamics plays a key role in both the characterization and design of new materials. The calixarenes have taken on a strong role in this area,1 complicated by the soft nature of the materials which allows guest driven solid–solid transformations and the existence of several different structures in the same single crystal. It is clear that additional characterization methods are very valuable in exploring such versatile materials. We show that for calixarenes, solid-state 1H NMR, in combination with ab initio calculations, can be useful for probing host–guest structure and dynamics.
Solid-state 1H NMR is challenging due to the relatively poor spectral resolution arising from the narrow 1H chemical shift range and strong 1H–1H dipolar interactions present in most materials. Probes capable of achieving fast MAS conditions (∼35 kHz) and ultrafast MAS conditions (∼70 kHz) offer increased resolution by reducing line-broadening effects of 1H–1H dipolar interactions. Furthermore, performing experiments at ultrahigh-fields offers a further gain in spectral resolution since the chemical shifts (in Hz) scale linearly with magnetic field strength, while the 1H–1H dipolar interactions remain constant. In this work, we present 1H MAS NMR spectra obtained under fast MAS conditions (32 kHz) and at an ultrahigh magnetic field strength of 21.1 T.†
Fig. 1 presents 1H MAS NMR spectra of five different forms of p-tert-butylcalix[4]arene: two polymorphs of the guest-free form, two different host–guest complexes with toluene, and the complex with pyridine. The 1H MAS NMR spectrum of the low-density guest-free form2 (Fig. 1a) consists of relatively broad resonances arising from CH3, CH2, aromatic, and OH protons, as indicated. However, the high-density guest-free form3 has a dramatically different spectrum, with two of the tert-butyl methyl resonances shifted from approximately 1 ppm to 0.26 and −1.20 ppm. These changes in the spectrum are a consequence of the “self-included” packing scheme of the high density form in which one tert-butyl group occupies the cavity of a neighbouring molecule. The peak at −1.20 ppm can likely be attributed to the additional magnetic shielding that this tert-butyl group experiences in the calixarene cavity, while the peak at 0.26 ppm likely arises from the tert-butyl group that points directly towards one of the aromatic rings of a neighbouring cavity, but from the outside of the cavity.
![900 MHz 1H MAS NMR spectra obtained with 32 kHz MAS frequency (left) and partial structures (right) of p-tert-butylcalix[4]arene complexes: (a) low-density guest-free, (b) high-density guest-free, (c) 2 : 1 complex with toluene, (d) 1 : 1 complex with toluene, (e) 1 : 1 complex with pyridine. The chemical shifts of selected resonances are given in ppm.](/image/article/2008/CP/b805326j/b805326j-f1.gif) | ||
| Fig. 1 900 MHz 1H MAS NMR spectra† obtained with 32 kHz MAS frequency (left) and partial structures (right) of p-tert-butylcalix[4]arene complexes: (a) low-density guest-free, (b) high-density guest-free, (c) 2 : 1 complex with toluene, (d) 1 : 1 complex with toluene, (e) 1 : 1 complex with pyridine. The chemical shifts of selected resonances are given in ppm. | ||
The 1H MAS NMR spectra for the calixarene host–guest complexes show that the protons of the guest molecules experience significant complexation-induced shifts (CISs). We refer here to a CIS as the difference between the chemical shift of a particular nucleus in the solid-state host–guest complex and in the “free” molecule. Solution chemical shifts for the guest molecules were used as reference values in calculating the CISs rather than solid chemical shifts for the practical reason that the guest molecules are not solids at ambient conditions, but also to avoid the contributions to the chemical shifts by neighbouring molecules in the crystals.
Table 1 compares the 1H chemical shifts for the guest molecules in the calixarene host–guest complexes to those observed in solution. Considering the narrow 1H chemical shift range (about 20 ppm), these CISs of up 4 ppm are quite remarkable. The 13C resonances for the toluene methyl groups in the 1 : 1 and 2 : 1 calixarene complexes show CISs of about 5 ppm, but these are only small changes compared to the 13C chemical shift range of about 200 ppm. The 1H resonances of the host calixarene do not display significant changes due to the presence of the guest molecule. For example, in the 1 : 1 complex with toluene, one might expect the tert-butyl protons to experience an increased shielding due to their proximity to the toluene aromatic ring. However, since the tert-butyl groups are rotating, only a small proportion of these protons are near the toluene molecule at a given time and the effect is probably averaged out.
| Experimentala/ppm | Calculatedb/ppm | |||||
|---|---|---|---|---|---|---|
| Solution | Complex | CIS | Free | Complex | CIS | |
| a Chemical shifts are reported as ppm from TMS, the solution chemical shifts are those for residual 1H signals in deuterated solvents. b Calculated chemical shifts are reported as ppm from the value calculated for TMS after conversion from shielding values. | ||||||
| Toluene (2 : 1) | ||||||
| H2 | 6.98 | 5.10 | −1.88 | 7.36 | 5.62 | −1.79 |
| H3 | 7.09 | 4.54 | −2.46 | 7.41 | 5.40 | −2.12 |
| H4 | 7.00 | 2.96 | −4.13 | 7.52 | 3.56 | −3.80 |
| H5 | 2.09 | −1.41 | −3.50 | 2.50 | −1.48 | −3.75 |
| Toluene (1 : 1) | ||||||
| H2 | 6.98 | 5.46 | −1.52 | 7.36 | 5.97 | −1.39 |
| H3 | 7.09 | 6.40 | −0.69 | 7.41 | 7.02 | −0.39 |
| H4 | 7.00 | 6.40 | −0.60 | 7.52 | 6.83 | −0.69 |
| H5 | 2.09 | −1.76 | −3.85 | 2.50 | −1.87 | −4.37 |
| Pyridine (1 : 1) | ||||||
| H2 | 8.74 | 6.92 | −1.82 | 9.15 | 7.71 | −1.44 |
| H3 | 7.22 | 4.46 | −2.76 | 7.30 | 4.95 | −2.35 |
| H4 | 7.58 | 4.29 | −3.29 | 7.97 | 4.75 | −3.22 |
For the 2 : 1 toluene complex4 (Fig. 1c), in which toluene resides in a capsule formed by two calixarene molecules, all of the toluene protons experience significant additional magnetic shielding, particularly the methyl (H5) and para (H4) protons. For the 1 : 1 toluene complex5,6 (Fig. 1d), in which each host calixarene cavity is occupied by a toluene guest molecule, it is the toluene methyl protons that experience the largest CIS. For pyridine (Fig. 1e), which is located in the calixarene cavity with its nitrogen pointing away from the four-fold rotation axis of the cavity,7 significant CISs are observed for all protons, especially H4 and H3.
These CISs arise primarily from the magnetic shielding provided by the aromatic rings of the calixarene cavities. In order to gain a fuller understanding of this effect, a map of the CISs in the calixarene cavity was constructed by performing ab initio calculations‡ of the additional magnetic shielding experienced by the nucleus of a single He atom at various points within the calixarene cavity. These maps plot the difference in the magnetic shielding between the nucleus of a “free” He atom and the “complexed” He atom (an increase in magnetic shielding corresponds to a negative CIS). Two slices through this 3D CIS map are presented in Fig. 2. According to these maps, the greatest CISs (of up to −6 ppm) are experienced in the regions nearest the aromatic rings (Fig. 2a). The map in Fig. 2b shows that the CIS is at a maximum at a depth of 4 Å above the bottom of the cavity. A more sophisticated approach to constructing similar types of maps has been recently presented by Sebastiani and co-workers.8
![Complexation induced shift maps in the p-tert-butylcalix[4]arene cavity constructed from ab initio calculations of the additional magnetic shielding of the nucleus of a helium atom at various positions in the cavity (with respect to a free He atom). The dashed lines indicate the slices through which the two views are taken: (a) view from above the cavity and (b) view from the side of the cavity. The He atoms were positioned on a 3D grid in the cavity with grid-points separated by approximately 0.6 Å. The displayed maps have been smoothed by interpolation and positions less than the van der Waals radius from host atoms have been removed.](/image/article/2008/CP/b805326j/b805326j-f2.gif) | ||
| Fig. 2 Complexation induced shift maps in the p-tert-butylcalix[4]arene cavity constructed from ab initio calculations‡ of the additional magnetic shielding of the nucleus of a helium atom at various positions in the cavity (with respect to a free He atom). The dashed lines indicate the slices through which the two views are taken: (a) view from above the cavity and (b) view from the side of the cavity. The He atoms were positioned on a 3D grid in the cavity with grid-points separated by approximately 0.6 Å. The displayed maps have been smoothed by interpolation and positions less than the van der Waals radius from host atoms have been removed. | ||
These CIS maps provide a useful qualitative tool for understanding the experimentally observed 1H chemical shifts presented in Fig. 1 and Table 1. For example, these maps show that, for the high-density self-included guest-free form, the tert-butyl group that resides in the cavity of a neighbouring calixarene experiences a significant amount of additional magnetic shielding, explaining the observed change in the 1H chemical shift of the protons of this tert-butyl group from about 1.0 ppm to −1.2 ppm.
The CISs observed for the 1 : 1 complexes involving toluene and pyridine can be rationalized with these maps. Fig. 3 displays a superposition of the positions of the guest molecules (determined in XRD experiments) on slices through the 3D CIS maps. For the 1 : 1 toluene–calixarene complex, Fig. 3a shows that the methyl (H5) protons are located near the region of the map with the maximum CISs, the ortho (H2) protons are located just slightly above the cavity in a region of medium shifts, while the meta (H3) and para (H4) protons are located above the cavity in regions with small shifts. This qualitatively agrees with the experimentally observed CISs of −3.85 ppm (H5), −1.52 ppm (H2), −0.69 ppm (H3), and −0.60 ppm (H4). The CIS map can also be used to predict the CISs for known proton positions. It is known that the toluene molecule is dynamic, with methyl group rotation, two-fold ring flipping, and four-fold rotation between the four symmetry related positions in the calixarene cavity.6 If such motions are accounted for, the CIS map predicts CISs of −3.99 ppm (H5), −1.63 ppm (H2), −0.44 ppm (H3), and −0.39 ppm (H4), in good agreement with the experimental observations. For the pyridine–calixarene complex, Fig. 3b shows that H4 is near a region with high shifts, H3 flips between regions of high and medium CISs, while H2 flips between regions of medium and low CISs. This qualitatively agrees with the experimentally observed CISs of −3.29 ppm (H4), −2.76 ppm (H3), and −1.82 ppm (H2). By accounting for the pyridine motions,7 the CISs predicted from the map are −3.76 ppm (H4), −2.44 ppm (H3), and −1.19 ppm (H4).
![Superposition of (a) toluene and (b) pyridine guest molecules on the complexation-induced shift map of the p-tert-butylcalix[4]arene cavity. The slices through the 3D CIS maps are taken such that they are very close to the ring planes of the guest molecules.](/image/article/2008/CP/b805326j/b805326j-f3.gif) | ||
| Fig. 3 Superposition of (a) toluene and (b) pyridine guest molecules on the complexation-induced shift map of the p-tert-butylcalix[4]arene cavity. The slices through the 3D CIS maps are taken such that they are very close to the ring planes of the guest molecules. | ||
Although a CIS map was not constructed for the capsule formed by two calixarene molecules in the 2 : 1 complex, the map constructed for a single cavity still provides a useful tool for explaining the observed CISs. The crystal structure of the 2 : 1 toluene complex4 reveals that the toluene methyl protons (H5) and the paraprotons (H4) are located nearest the regions of the cavities that give the largest CISs. Indeed, this is what is observed experimentally. The other protons (H2 and H3) experience notable shifts as well, likely a consequence of these protons experiencing the shielding effects of two calixarenes, rather than just one.
Ab initio calculations of the guest molecule 1H chemical shifts‡ were also carried out using the structural coordinates provided by the single crystal XRD structures of the various complexes. For the 1 : 1 complexes, the calculations were performed on the complex of one guest molecule with one calixarene extracted from the extended crystal structures, as depicted in Fig. 1d and e. For the 2 : 1 complex, the calculations were performed on a single capsule extracted from the crystal structure, consisting of two calixarenes and one toluene guest molecule (Fig. 1c). The positions of all protons of the host and guest molecules were optimized prior to calculation of NMR chemical shifts. The calculated shifts were averaged to give a single value for the protons known to exchange with each other due to the motions of the guest molecules. The ab initio calculated 1H chemical shifts of the guest molecule protons in both the free and complexed forms are provided in Table 1. Although the absolute values for the calculated 1H chemical shifts tend to overestimate the experimental shifts, the calculated CISs are in excellent agreement with experiment.
Due to the large number of atoms in the unit cells, it was not possible for us to perform calculations that take into account the periodicity of the structure.9 Nonetheless, it has been possible to accurately calculate the 1H chemical shifts using isolated pairs of guest and host molecules, rather than the extended crystalline environment. This success can be primarily attributed to the fact that the magnetic shielding effects that give rise to these CISs are mainly localized to within a single calixarene cavity (with the exception of the high density self-included guest-free form).
The guest molecule CISs predicted from the maps and calculated with ab initio methods are both in good agreement with the experimental data. While this is a satisfactory result in itself, it is important to point out that the structures and dynamics of the host–guest complexes were known ahead of time. Would it be possible to provide structural information about an unknown structure from the measurement and calculations of these CISs?
Consider the 1 : 1 calixarene–toluene complex: is it possible to determine the toluene location in the calixarene cavity from the observed 1H chemical shifts and the calculated CIS maps in Fig. 2? Given the large CIS of the methyl group observed experimentally, it is obvious that the methyl group must point into the cavity, rather than out, and be located near the regions shown by the map to have large shifts. How deep is the toluene molecule inserted into the cavity? To try and answer this, the toluene molecule was aligned with the four-fold rotation axis of the calixarene with the methyl group pointing into the cavity, and was moved along the z-direction. The sum of squares of the difference (χ2) between the experimental CISs and those calculated from the CIS maps was calculated for each position. Fig. 4a displays this χ2 value as a function of distance between the center of the toluene ring and the bottom of the calixarene cavity. There is a clear minimum at approximately 6.6 Å, in exact agreement with the toluene position determined by XRD.5Fig. 4b shows that the χ2 value is rather insensitive to rotation of the toluene molecule around the z-axis. The minimum is located at 0° whereas the single crystal XRD structure has the toluene molecules rotated by about 20°. The toluene molecules are actually located slightly off the four-fold rotation axis and are tilted away slightly from this axis (see Fig. 3a), but χ2 is not sensitive enough to these small deviations.
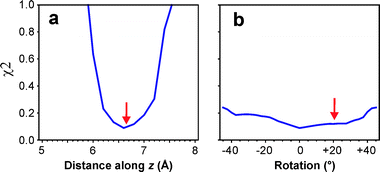 | ||
| Fig. 4 Sum of squares difference (χ2) between experimental 1H CISs of the 1 : 1 toluene–calixarene complex and CISs calculated from the CIS maps as a function of (a) distance between centre of toluene ring and bottom of calixarene cavity along the z axis (four-fold rotation axis) with rotation fixed at 0° and (b) rotation of toluene molecule about the z axis with position fixed at 6.6 Å above the bottom of the cavity. The rotation angle refers to the clockwise rotation of the plane of the toluene ring away from the plane defined by the dashed line in Fig. 2a. The arrows indicate the position of the toluene molecule in the single-crystal XRD structure. | ||
The general location of the toluene molecules in the calixarene cavities can be determined using this approach, illustrating that useful structural information is available in the measurement and calculation of 1H CISs, demonstrating the potential for using chemical shifts for NMR crystallography of host–guest complexes. These data can complement other NMR data such as 2H NMR that provides information about guest molecule dynamics.6,7 As is the case for many other systems, each NMR method is best used in combination with other NMR techniques, other experimental characterization techniques, and computational methods in order to obtain a greater understanding of the structure of interest.
References
- J. A. Ripmeester, G. D. Enright, C. I. Ratcliffe, K. A. Udachin and I. L. Moudrakovski, Chem. Commun., 2006, 4986 RSC; S. J. Dalgarno, P. K. Thallapally, L. J. Barbour and J. L. Atwood, Chem. Soc. Rev., 2007, 36, 236 RSC; A. W. Coleman, S. Jebors, P. Shahgaldian, G. Ananchenko and J. A. Ripmeester, Chem. Commun., 2008, 2291 RSC.
- G. D. Enright, K. A. Udachin, I. L. Moudrakovski and J. A. Ripmeester, J. Am. Chem. Soc., 2003, 125, 9896 CrossRef CAS; E. B. Brouwer, K. A. Udachin, S. Lang, K. Ooms, P. A. Halchuk and J. A. Ripmeester, Chem. Commun., 2003, 1416 RSC; J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottle, Science, 2002, 298, 1000 CrossRef CAS.
- E. B. Brouwer, K. A. Udachin, G. D. Enright, J. A. Ripmeester, K. J. Ooms and P. A. Halchuk, Chem. Commun., 2001, 565 RSC.
- G. D. Enright, E. B. Brouwer, P. A. Halchuk, K. J. Ooms, M. J. Ferguson, K. A. Udachin and J. A. Ripmeester, Acta Crystallogr., Sect. A, 2002, 58, C310 CrossRef.
- G. D. Enright, E. B. Brouwer, K. A. Udachin, C. I. Ratcliffe and J. A. Ripmeester, Acta Crystallogr., Sect. B, 2002, 58, 1032 CrossRef.
- E. B. Brouwer, G. D. Enright, C. I. Ratcliffe and J. A. Ripmeester, Supramol. Chem., 1996, 7, 79 CrossRef CAS.
- E. B. Brouwer, G. D. Enright, C. I. Ratcliffe, G. A. Facey and J. A. Ripmeester, J. Phys. Chem. B, 1999, 103, 10604 CrossRef CAS.
- D. Sebastiani, ChemPhysChem, 2006, 7, 164 CrossRef CAS , and references therein; G. Brunklaus, A. Koch, D. Sebastiani and H. W. Spiess, Phys. Chem. Chem. Phys., 2007, 9, 4545 Search PubMed.
- D. Sebastiani and M. Parrinello, J. Phys. Chem. A, 2001, 105, 1951 CrossRef CAS; S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CrossRef CAS.
Footnotes |
| † Access to the 900 MHz NMR spectrometer was provided by the National Ultrahigh-Field NMR Facility for Solids (www.nmr900.ca). |
| ‡ Ab initio calculations were performed with Gaussian 98 revision A.11.3 (Gaussian, Inc., Pittsburgh PA, 2002). Proton geometry optimizations were performed at the B3LYP/6-31G or B3LYP/6-31G(d) level and NMR calculations employed the GIAO method at the HF/6-31G(d,p) level. |
| This journal is © the Owner Societies 2008 |