Neomycin-capped aromatic platforms: quadruplex DNA recognition and telomerase inhibition
Markus
Kaiser†
a,
Anne
De Cian
b,
Matthieu
Sainlos
a,
Christian
Renner
c,
Jean-Louis
Mergny
b and
Marie-Paule
Teulade-Fichou
*a
aLaboratoire de Chimie des Interactions Moléculaires, Collège de France, CNRS UPR 285, 11, place Marcelin Berthelot, 75005 Paris, France. E-mail: mp.teulade-fichou@college-de-france.fr; Fax: ++33 (0) 1 44 27 13 56
bLaboratoire de Biophysique, Muséum National d'Histoire Naturelle USM 503, INSERM U 565, CNRS UMR 5153, 43 rue Cuvier, 75005 Paris, France
cSchool of Biomedical and Natural Sciences, Nottingham Trent University, Clifton Lane, Nottingham, United Kingdom NG11 8NS
First published on 25th January 2006
Abstract
A series of aminoglycoside-capped macrocyclic structures 9–12 has been prepared using intramolecular bis-tethering of neomycin on three aromatic platforms (phenanthroline, acridine, quinacridine). Based on NMR and calculations studies, it was found that the cyclic compounds adopt a highly flexible structure without conformational restriction of the aminoglycoside moiety. FRET-melting stabilization measurements showed that the series displays moderate to high affinity for the G4-conformation of human telomeric repeats, this effect being correlated with the size of the aromatic moiety. In addition, a FRET competition assay evidenced the poor binding ability of all macrocycles for duplex DNA and a clear binding preference for loop-containing intramolecular G4 structures compared to tetramolecular parallel G4 DNA. Finally, TRAP experiments demonstrated that the best G4-binder (quinacridine 11) is also a potent and selective telomerase inhibitor with an IC50 in the submicromolar range (200 nM).
Introduction
Inhibition of telomerase or alteration of the telomere state are both valuable concepts for inducing senescence and apoptosis in cancer cells.1 A simultaneous targeting of telomeres and the nucleoenzyme telomerase was recently demonstrated as a promising approach for limiting cancer cell proliferation.2 The two processes are intimately connected: modulation of the telomeric structure impairs telomerase binding, resulting in inactivation of both the catalytic and the maintenance activities of the enzyme.3 Thus, compounds able to disrupt the telomeric structure are particularly interesting as potential telomere function modulators and telomerase inhibitors.4 Structural perturbation of the telomeres can be reached by inducing a folding of the G-rich telomeric 3′ overhang into a quadruplex conformation, which can be achieved by binding of a highly selective G-quadruplex ligand.4,5 Therefore, a reasonable strategy for identifying novel anticancer drugs relies on the discovery of strong and selective G4 ligands with consequent telomerase inhibition.6 In the past decade, thousands of compounds were screened according to this approach and three main classes of ligands could be established: i) fused polycyclic intercalators,7 ii) macrocyclic compounds of either natural or synthetic origin,8 and iii) polyaromatic unfused systems.6a,9 However, structural data on the molecular interactions between the ligands and their G4-DNA target are still scarce and no general concept for the design of highly selective binders is available. In spite of several structural studies which provided valuable information about the interactions of small molecules with loops,10a–f a deeper understanding is still needed, and in particular little is known about groove occupancy.10g,h Additionally, folded quadruplexes show a highly dynamic and polymorphic structure,11 further complicating the drug design. As a consequence, the search for G4-binders of high specificity has been proved difficult and remains a challenging task.In the course of our studies on the design of G4-binders, we developed the pentacyclic crescent-shaped quinacridine motif that shows a high affinity for quadruplex DNA mainly due to strong stacking interactions with G-quartets.7c,d Moreover, a dimeric macrocyclic bisquinacridine was shown to elicit a high preference for quadruplex over duplex DNA.8b Macrocyclic scaffolds are particularly attractive for designing G4 ligands, as they show a preferential binding to “non-standard” DNA conformations, due to their sterically difficult intercalation between the base pairs of “standard” B-DNA.12 In contrast, the external G-quartets of quadruplexes constitute accessible planar sites of large area which can accommodate large-sized molecules. This is remarkably illustrated by the exquisite G4-binding specificity of the natural compound telomestatin which is composed of seven oxazole rings and a dihydro-thiazole moiety combined in a cyclic scaffold.8a,13
Aminoglycosides are natural antibiotics which have been widely used to achieve selective recognition of various loop or bulge-containing RNA structures.14 Despite exhibiting a low affinity towards G4-quadruplex structures per se,15 aminoglycosides possess several ammonium centers able to establish multiple salt bridges and H-bonding contacts with nucleic acids. In particular neomycin has been shown to exhibit a high selectivity for DNA triplexes.16 In addition, the 1,3-hydroxylamine motif commonly found in aminoglycosides has been identified as a recognition motif for the complexation of phosphate groups and of the Hoogsteen face of guanine via a bidentate H-bonding/electrostatic interaction.17 Altogether, and in view of the presence of short loops in intramolecular G4 structures, we speculated that an appropriate derivatization of aminoglycosides with an intercalator system could lead to high-affinity ligands through simultaneous targeting of the G-quartet surface and of the loops phosphate or residues. Along this line, we decided to assemble the two motifs in a cyclic scaffold in the hope of compromising the duplex binding. Hence, capping of various aromatic platforms with a neomycin moiety has been investigated as an approach to the design of cyclic “dome-shaped” scaffolds suitable for adapting the topology of loop-containing quadruplexes.
Results and discussion
Synthesis
Condensation of a dialdehyde with a diamine is a well-known strategy for constructing macrocyclic structures.18 This approach has proven to be very high-yielding if performed under moderate to high dilution conditions for minimising unwanted polymerisation. The aminoglycoside neomycin features two reactive primary amino groups linked to a methylene carbon whereas its other amines are directly linked to sugar units and are thus sterically less accessible for derivatization. Based on this structural peculiarity, regioselective protection of the amino groups of neomycin can be achieved via a benzoylation–Teoc-protection–hydrogenation sequence (Scheme 1).19 Derivatization of the neomycin intermediate 4 with a Boc-protected lysine or aminocaproic acid building block, led after deprotection to elongated neomycin diamine derivatives 7 and 8, respectively. These were then condensed with three different dicarboxaldehyde aromatic platforms (acridine, quinacridine, phenanthroline) by reductive amination, yielding the four macrocycles 9–12. Although Schiff base macrocyclisation can lead to the formation of either [1 + 1] or [2 + 2] coupling products,20 in our case the predominance of a [1 + 1] condensation of the partners was evidenced by HPLC analysis, and appears independent of the size of the aromatic system. A reasonable explanation for the predominance of the intramolecular cyclisation is that pre-organization of the derivatized neomycin moiety occurs, to a certain extent, during tethering. Although the aminoglycoside scaffold is flexible, it is prone to adopt defined conformations upon limitation of the rotation of the sugar units. Therefore, it can be hypothesized that the aminoglycoside moiety, after the first amination, adopts a conformation placing the reactive groups favourably for internal ring closure. Hence, entropic contribution due to optimal spatial fitting of the amino and aldehyde functions, along with high dilution conditions, could both participate to the predominance of the [1 + 1] coupling.![Reagents and conditions: (a) N-(Z)-5-norbornene-2,3-dicarboximide, TEA, DMSO–H2O (10 : 1), rt, 12 h, 51%; (b) Teoc-p-nitrophenyl carbonate, TEA, dioxane–H2O (3 : 1), 55 °C, 48 h, 87%; (c) H2, Pd/C (10%), MeOH–H2O (9 : 1), rt, 2 h, 81%; (d) EDCI/HOAt, TEA, DMF and Nα-(Boc)–Lys(Z)–OH or N-(Z)-capronic acid, rt, 12 h, 92% for 5, 86% for 6; (e) H2, Pd/C (10%), MeOH–H2O (9 : 1), rt, 2 h, 92% for 7, 94% for 8; (f) i) TEA, DCM–MeOH (1 : 1) and 2,5-bis(dicarboxaldehyde)acridine or dibenzo[b,j][1,7]phenanthroline-2,10-dicarboxaldehyde or 2,9-bis(dicarboxaldehyde)-1,10-phenanthroline, rt, 4 d; ii) NaBH4, DCM–MeOH (1 : 1), rt, 2 h; iii) TFA–DCM (1 : 1), rt, 1 h, 29% for 9, 42% for 10, 37% for 11, 12% for 12.](/image/article/2006/OB/b516378a/b516378a-s1.gif) | ||
| Scheme 1 Reagents and conditions: (a) N-(Z)-5-norbornene-2,3-dicarboximide, TEA, DMSO–H2O (10 : 1), rt, 12 h, 51%; (b) Teoc-p-nitrophenyl carbonate, TEA, dioxane–H2O (3 : 1), 55 °C, 48 h, 87%; (c) H2, Pd/C (10%), MeOH–H2O (9 : 1), rt, 2 h, 81%; (d) EDCI/HOAt, TEA, DMF and Nα-(Boc)–Lys(Z)–OH or N-(Z)-capronic acid, rt, 12 h, 92% for 5, 86% for 6; (e) H2, Pd/C (10%), MeOH–H2O (9 : 1), rt, 2 h, 92% for 7, 94% for 8; (f) i) TEA, DCM–MeOH (1 : 1) and 2,5-bis(dicarboxaldehyde)acridine or dibenzo[b,j][1,7]phenanthroline-2,10-dicarboxaldehyde or 2,9-bis(dicarboxaldehyde)-1,10-phenanthroline, rt, 4 d; ii) NaBH4, DCM–MeOH (1 : 1), rt, 2 h; iii) TFA–DCM (1 : 1), rt, 1 h, 29% for 9, 42% for 10, 37% for 11, 12% for 12. | ||
NMR studies and calculations
In order to gain insight into the conformational flexibility of the final capped macrocycles, the acridine derivative 9 was studied by NMR. Two relatively well-defined conformers are found for the pucker and relative orientations of the sugar rings (Fig. 1). In one conformation the overall shape of the tetrasaccharide resembles that of an “L” in which sugars B, C and D are extended and form the long stem of the “L”. In the other conformation rings A, B and C are arranged in a helix-like manner leading to a V-shaped appearance. The aliphatic chain of Lys-1 packs in both cases against sugar A, but the aromatic moiety and Lys-2 exhibit variable orientations and seem quite flexible. The fact that only one set of NMR resonances was observed indicates fast conformational averaging on the NMR time-scale of milliseconds. The two distinct conformations were detected in the initial structure calculations with all NMR-derived distance constraints. Because each conformation showed systematic and characteristic violation of a few NMR distances, subsets of the NMR constraints could be derived that corresponded exactly to the two conformers. All NMR structures are quite compact with the aromatic moiety fairly close to the sugar rings. In solution, however, the molecules might also partly exist in more open forms that cannot be detected by NMR due to the bias of the Overhauser effect towards short distances and, therefore, compact structures.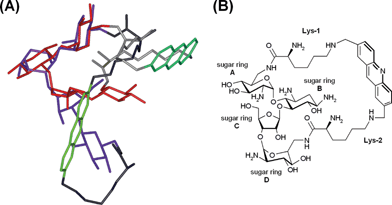 | ||
| Fig. 1 (A) Superimposition of the two solvent conformations of 9 as derived from NMR analysis (acridine system: light and dark green, lysine linker: light and dark grey, neomycin moiety: blue, L shaped and red, V-shaped). (B) Nomenclature of the structural elements of 9. | ||
In conclusion, although the incorporation of aromatic systems could have been expected to cause rigidity, rotation of the sugar moieties in the final macrocycle seems not to be restrained by the double tethering. This is likely attributable to the length and flexibility of the lysine linkers. Thereby, compound 9 appears to be a highly adaptable structure, and it is reasonable to assume that macrocycles 10–12 adopt identical conformational behaviours.
FRET-melting stabilization assay
The interaction of the macrocycles with quadruplex DNA has been investigated by a high-throughput FRET assay using a real-time PCR apparatus and the doubly labelled F21T oligonucleotide which mimics the human telomeric single-strand overhang.21 ΔT1/2 values were concentration dependent; as shown in Fig. 2. Moderate to high stabilizations of F21T were observed in the presence of macrocycles 9–12 at 0.2–5 µM (Fig. 2). Interestingly, this effect is correlated with the size of the aromatic moiety since the most active compound is the quinacridine derivative 11 (ΔT1/2 = 14.1 ± 1.1 °C at 1 µM), whereas the acridine and phenanthroline derivatives 10 and 12 show a much lower activity (ΔT1/2 = 6.3 and 6.0 °C at 1 µM, respectively). Even at submicromolar concentrations (0.2–0.5 µM), a significant stabilization is found with compounds 9 and 11 (Fig. 2). As the three ligands 10–12 exhibit the same cationic charge, this result evidences the strong π-stacking contribution to the binding since extension of the surface area in contact is known to increase the attraction between aromatic systems.22 The importance of electrostatics in the stabilization of the G4 conformation is illustrated by the higher effect of the acridine derivative 9 which bears two more cationic amino groups than its analogue 10 [Δ(ΔT1/2)9–10 = + 4.8 °C]. Importantly, free neomycin has little or no effect on the melting of F21T (ΔT1/2 = 2.1 °C at the highest drug concentration tested, 5 µM), which reveals that the high cationic charge (4+–6+) of the free aminoglycoside23 is not sufficient to ensure an efficient binding of the G4 structure. In addition, the acyclic tetraamino phenanthroline derivative MOP1 (Fig. 3) has a very limited effect on the melting (ΔT1/2 = 1.4 and 3.5 °C at 1 and 5 µM, respectively) and the diamino acridine MonoA was previously found to be completely ineffective (ΔT1/2 = 0 °C at 1 µM).24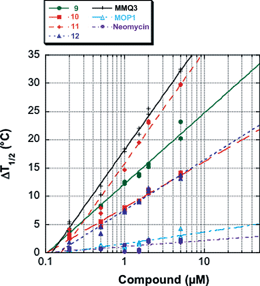 | ||
| Fig. 2 Concentration dependency of F21T stabilization (ΔT1/2 values, in °C). Data is presented on a semi-log scale for MMQ3 (crosses), 11 (orange diamonds), 9 (green circles), 10 (red squares), 12 (blue triangles), MOP1 (light blue triangles) and neomycin (purple circles). | ||
 | ||
| Fig. 3 Structures of the acyclic control compounds. | ||
Taken together, these data demonstrate that a synergistic effect is obtained in combining the aromatic and aminoglycoside motifs in the same scaffold, at least in the phenanthroline and acridine series. Indeed, the additivity of the aromatic and electrostatic contributions is not so obvious in the quinacridine series as the acyclic control MMQ3 and ligand 11 display similar ΔT1/2 values at all concentrations (Fig. 2). This might indicate that strong π-stacking of the quinacridine moiety masks the other effects, or that the relative weights of the various energetic contributions differ for the two ligands.
Competitive FRET-melting assay
A high binding selectivity is an essential criterion for the use of G4-binders in complex environments. Hence the G4-selectivities of our macrocycles were evaluated using a competitive FRET-melting assay. To this end melting of F21T is performed in the presence of various DNA competitors: a 26bp duplex (ds26) as described previously,7c and two quadruplexes containing 5 guanine quartets: a four-stranded parallel quadruplex [TG5T]4 and an intramolecular quadruplex (30AG) (Fig. 4).![Selectivity in FRET assay. Thermal denaturation of F21T was performed in the presence of the various compounds (1 µM) and in the presence of various competitors: (A) double-stranded DNA ds26, (B) parallel tetramolecular G-quadruplex [TG5T]4, (C) intramolecular G-quadruplex 30AG. The stabilization (ΔT1/2) is reported in °C for each compound and the following concentrations of competitors: no competitor; ds26 3 µM (dark blue), 10 µM (light blue); (TG5T)4 1 µM (dark green), 3 µM (light green); 30AG 1 µM (dark red), 3 µM (light red). (D) Apparent melting temperature (T1/2) of F21T in the presence of 1 µM of compound 11 with various concentrations of competitors (ds26 duplex: filled circles; 30AG: open cicles and [TG5T]4: crosses). (E) Apparent melting temperature (T1/2) of F21T in the presence of 1 µM of compound MMQ3 with various concentrations of competitors (ds26 duplex: filled circles; 30AG: open circles and [TG5T]4: crosses).](/image/article/2006/OB/b516378a/b516378a-f4.gif) | ||
| Fig. 4 Selectivity in FRET assay. Thermal denaturation of F21T was performed in the presence of the various compounds (1 µM) and in the presence of various competitors: (A) double-stranded DNA ds26, (B) parallel tetramolecular G-quadruplex [TG5T]4, (C) intramolecular G-quadruplex 30AG. The stabilization (ΔT1/2) is reported in °C for each compound and the following concentrations of competitors: no competitor; ds26 3 µM (dark blue), 10 µM (light blue); (TG5T)4 1 µM (dark green), 3 µM (light green); 30AG 1 µM (dark red), 3 µM (light red). (D) Apparent melting temperature (T1/2) of F21T in the presence of 1 µM of compound 11 with various concentrations of competitors (ds26 duplex: filled circles; 30AG: open cicles and [TG5T]4: crosses). (E) Apparent melting temperature (T1/2) of F21T in the presence of 1 µM of compound MMQ3 with various concentrations of competitors (ds26 duplex: filled circles; 30AG: open circles and [TG5T]4: crosses). | ||
As shown in Fig. 4A, the four macrocycles display a good selectivity for the quadruplex over the control duplex, since stabilization of F21T is only moderately affected at the highest concentration of the duplex (10 µM). It is worth pointing out that this is a stringent competition in terms of electrostatics and quartet vs. base-pair binding, since ds26 is added in large excess to F21T (respectively 15 and 50 molar eq.). On the other hand, MMQ3 is displaced more easily from F21T, as seen from the strong drop in ΔT1/2 observed even at the lowest concentration of the duplex competitor (3 µM), reflecting likely intercalation of the acyclic compound into the duplex DNA. In turn, the ability of the macrocyclic series to distinguish between the quadruplex and the duplex might be attributed to a poor insertion into double-stranded DNA, supporting our initial design to achieve quadruplex vs. duplex selectivity by cyclisation.
In order to evaluate the possible interaction of the macrocyclic compounds with the TTA loops of the G-quadruplexes, we compared the decrease of ΔT1/2 obtained with the ds26 duplex and the two types of G4 structure: the tetramolecular [TG5T]4 quadruplex, which does not contain any loop, and the intramolecular G4 formed by 30AG. When pre-formed in favourable conditions, [TG5T]4 cannot be denaturated in the conditions of the FRET assay25 whereas ds26 and 30AG have a Tm of 70.5 °C and 74 °C, respectively, under identical conditions (Na cacodylate 10 mM pH 7.2, NaCl 100 mM) (data not shown). Melting of the 30AG is dependent on the cation and the signature of the thermal difference spectrum is consistent with a G-quadruplex.26 Despite the lack of data on the exact structure or on the loop conformation, it is likely that this oligonucleotide adopts a structure close to that of F21T. Note that it is important to select competitors which are thermally more stable that the F21T quadruplex; otherwise they would be unfolded in the temperature range chosen for the melting studies, and would therefore act as single-stranded rather than double-stranded or quadruple-stranded competitors.
When the competition assay is carried out in the presence of 1 and 3 µM of the oligonucleotides forming quadruplex structures (respectively 5 and 15 molar equivalents compared to F21T but only 1 and 3 molar equivalents compared to the ligand), (Fig. 4B,C) the drop in ΔT1/2 for the macrocyclic series is more pronounced than for the duplex competitor. The comparison of the destabilization obtained when adding 3 µM of each competitor results in a decrease of 14 to 40% with ds26, of 53 to 73% with [TG5T]4 and the stabilization is fully abolished with 30AG. Interestingly, the results are somewhat different for MMQ3 as the addition of 3 µM of ds26 results in a decrease in ΔT1/2 of 12.2 °C (80%), whereas the addition of the same amount of G4 structure [TG5T]4 leads to a decrease of only 11 °C (72%). This is again quite consistent with the role of the neomycin capping in the preference of the macrocyclic series for quadruplex over duplex DNA.
For [TG5T]4 at least 30% of the stabilization of F21T is maintained in all cases, at the two concentrations of competitor used. This result suggests that all the ligands tested exhibit a higher affinity for the intramolecular quadruplex conformation of F21T compared to the tetramolecular form of [TG5T]4. In contrast, when adding the intramolecular G4 30AG, the competition pattern shows a strong decrease in ΔT1/2 for all compounds and even a complete loss of stabilization when 30AG is used at 3 µM concentration (3 molar equivalent compared to ligand) (Fig. 4C). The comparison with the competitor [TG5T]4 suggests that these ligands are more easily displaced from F21T when the competitor G4-DNA contains loops. Interestingly, MMQ3 seems to be slightly less sensitive than 11 to the G4 competitor with loops, mostly at 1 µM, and as emphasized earlier, it has more affinity for the tetramolecular quadruplex than 11. It is worth noting that the complex with the compound 11 is clearly less affected by the tetramolecular G4 or the ds26 duplex (Fig. 4D) than the complexes with the control MMQ3 (Fig. 4E).
All together these data demonstrate that the neomycin-capped macrocycles and in particular macrocycle 11 are able to discriminate between loop-containing and tetramolecular parallel quadruplexes, strongly suggesting a possible interaction of the small-molecule binders with the loops. The presence of loops may contribute to the binding via direct contacts with the ligands or indirectly via conformational constraint of the target. Quadruplex/duplex selectivity has been estimated using two independent methods:
● ΔT1/2 values in the presence of increasing concentrations of duplexes or quadruplexes unambiguously indicate that an intramolecular quadruplex (30AG) is a much better competitor than a self-complementary duplex (ds26) or a parallel quadruplex [TG5T]4 (Fig. 4D). The addition of 1.1 µM 30AG leads to a 50% decrease in ΔT1/2, whereas >5 µM ds26 is required to obtain the same ΔT1/2. These two-strand concentrations correspond to 5.5 µM quartets and 71 µM base pairs, respectively, demonstrating that a much larger molar excess of base pairs is required to abolish half of the ligand-induced stabilization of F21T. Even at 30 µM, ds26 does not totally abolish stabilization by compound 11, in constrast with 30AG, for which stabilization is completely lost at 10 µM.
● An equilibrium dialysis experiment using a limited set of compounds confirms a preference for 30AG over duplexes and parallel quadruplexes (data not shown).
In other words, both methods confirm that 11 has a preference for intramolecular quadruplexes over tetramolecular quadruplexes and duplexes, in contrast with MMQ3 which exhibits little, if any, selectivity (Fig. 4E).
Telomerase inhibition
Finally the compounds were examined for their ability to inhibit human telomerase activity in vitro. Inhibition of telomerase was measured by the TRAP assay with an internal standard to ensure the validity of the test.27 The assay was performed at increasing ligand concentrations; analysis by denaturating gel electrophoresis is shown in Fig. 5A and quantitative analysis is provided in Fig. 5B. As shown on the gel, the IC50 values of the macrocycles rank in the low micromolar to submicromolar range, the most potent effect being observed for compound 11 with an IC50 of 0.2 µM (Fig. 5C). In all cases, the internal control (ITAS) is only affected at significantly higher concentrations compared to the IC50, in agreement with the selective binding of the compounds. Finally, it is worth noting that the IC50 values correlate well with the G4-stabilization effects (ΔT1/2) determined by the FRET-melting assay, again supporting the validity of the inhibition strategy based on the conformational modification of the substrate DNA.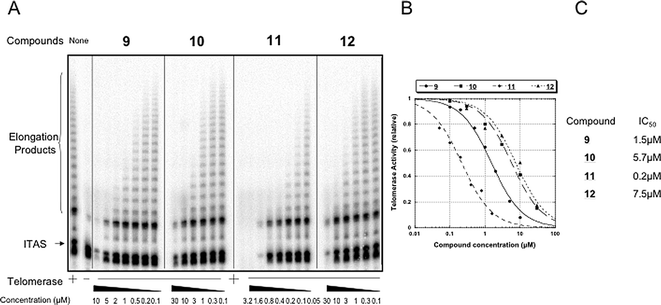 | ||
| Fig. 5 Telomerase inhibition by compounds 9–12 in a TRAP assay. (A) Increasing concentrations of compounds were added to the TRAP mixture in the presence of an internal control (ITAS) and analysed by gel electrophoresis. TRAP activity was determined with 200 ng of a CHAPS extract of the A431 cell line. IC50s were determined by comparison with the telomerase activity with no compound. They are given with a 20% precision. (B) Quantitative analysis of telomerase activity as a function of drug concentration. (C) IC50 values against telomerase found for the different compounds. | ||
Conclusion
The macrocyclic series described in the present study exhibits a good to high affinity for intramolecular quadruplexes and a good selectivity for DNA quadruplexes vs. duplexes. Given the poor affinity and selectivity of the acyclic controls, the G4 preferential binding of the macrocycles could be mediated by their particular cyclic conformation. Moreover, the synergistic effects obtained in the acridine and phenanthroline series strongly suggest that the neomycin motif is likely to play a role in establishing specific contacts with the G4-DNA target.In addition, the competition assay with a parallel tetramolecular quadruplex established that our compounds preferentially interact with loop-containing quadruplexes. This suggests the involvement of loop motifs in the binding of the macrocycles, confirming the structural importance of the loop for G4-DNA recognition. Macrocycles 9–12 display sterically hindered conformations but elicit a high plasticity, therefore, it would be of great interest to further investigate their binding mode for understanding how they adapt to the compact structure of G-quadruplex DNA, and whether they are able to recognize the conformational diversity of intramolecular quadruplexes.11g
Finally the TRAP assay demonstrated that macrocycle 11 inhibits telomerase in vitro in the submicromolar range, and thus could serve as a lead for the design of a new series able to act as telomere maintenance modulators. In addition, the concept of G4 sequestration of telomeric DNA by small molecules holds promise for interfering with other telomere binding proteins such as POT1, which actively participates in the prevention of the unfolding of the 3′-overhang and was recently shown to disrupt G4-quadruplexes.28
In conclusion, our neomycin-capped macrocycles display two interesting features (i.e. quadruplex recognition and telomerase inhibition) making them attractive structural scaffolds for further developments aiming at the discovery of new and more selective anticancer agents.
Experimental
General methods
All commercially available chemicals were reagent grade and were used without further purification. Flash chromatography employed Merck silica gel [Kieselgel 60 (0.040–0.063 mm)]. Analytical TLC was performed with 0.2 mm silica-coated aluminium sheets, visualization by UV light or by spraying either a solution of ninhydrin (0.3% in weight in n-butanol containing 3% acetic acid in volume) or an iodine solution (0.1 M in 10% sulfuric acid aqueous solution). Preparative reversed-phase HPLC was carried out on an Abimed-Gilson chromatograph using a Nucleodur 100 C18 ED 5µ (250 × 10 mm) (Macherey & Nagel, Düren, Germany) and gradients from 0.1% aqueous TFA to CH3CN containing 0.1% TFA (flow rate: 3 mL min−1).1H and 13C NMR spectra were recorded on a Bruker Avance300 spectrometer. For 1H and 13C, chemical shifts are reported in ppm (δ) downfield of tetramethylsilane (TMS) used as internal standard.
LC HR MS measurements were performed on an Agilent 1100 series HPLC with an XTerra MS C8 3.5 µm column (2.1 × 100 mm column dimension) coupled to a Bruker Daltonik microTOF mass spectrometer (electrospray ionisation). The following gradient was used: 5% aq. acetonitrile (0.05% trifluoroacetic acid) to 90% aq. acetonitrile (0.05% trifluoroacetic acid) in 15 min at a flow of 250 µL min−1.
The three aromatic dialdehyde building blocks have already been described29 and the synthesis of compounds 2–8 will be reported elsewhere.
NMR analysis
NMR spectra were recorded at 10 °C on a Bruker DRX 500 and an AV900 spectrometer equipped with pulsed-field-gradient (PFG) accessories and a cryoprobe in the case of the 900 MHz spectrometer. The sample was dissolved in a 9 : 1 H2O–D2O (v/v) mixture or in >99.9% D2O at 5 mM concentrations resulting in an (uncorrected) pH of 4. Resonance assignments were performed according to the method of Wüthrich.30 The 2D TOCSY was recorded with a spin-lock period of 70 ms using the MLEV-17 sequence for isotropic mixing.31 The 49 experimental distance constraints were extracted from 2D NOESY32 and ROESY33 experiments with mixing times of 200 ms. Water suppression was achieved with the WATERGATE scheme34 for samples containing 90% H2O and via presaturation in the case of D2O.Structure calculation
Structure calculations and evaluations were performed with the INSIGHT II 2000 software package (Accelrys, San Diego, CA) on Silicon Graphics O2 R5000 computers (SGI, Mountain View, CA). A hundred structures were generated from the distance-bound matrices. Triangle-bound smoothing was used. The NOE intensities were converted into interproton distance constraints using the following classification: very strong (vs) 1.7–2.3 Å, strong (s) 2.2–2.8 Å, medium (m) 2.6–3.4 Å, weak (w) 3.0–4.0 Å, very weak (vw) 3.2–4.8 Å, and the distances of the pseudo atoms were corrected as described by Wüthrich.27 The structures were generated in four dimensions, then reduced to three dimensions with the EMBED algorithm and optimized with a simulated annealing step according to the standard protocol of the DG II package of INSIGHT II. All hundred structures were refined with a short MD-SA protocol: after an initial minimisation, 5 ps at 300 K were simulated followed by exponential cooling to ∼0 K during 10 ps. A time step of 1 fs was used with the CVFF force-field while simulating the solvent H2O with a dielectric constant of 80.0. The experimental distance constraints were applied at every stage of the calculation with 50 kcal mol−1 Å−2.After simulated annealing with DISCOVER the structures were sorted according to their final energies and the structures with the lowest energies were analyzed. In the first calculations with all 49 NMR-derived distance constraints two conformational families were obtained, both with four characteristic violations. Two subsets of NMR constraints were constructed by removing once the four constraints that were persistently violated in the first conformational family and once the other four constraints that could not be fulfilled by the second conformational family. All low-energy structures that resulted from calculations with one of the subsets belonged to the corresponding conformational family and exhibited no significant violations of distance constraints.
Oligonucleotides
All oligonucleotides were synthesized and purified by Eurogentec (Belgium). The parallel quadruplex (TGGGGGT)4 was obtained after incubation of the monomer at 500 µM in a 10 mM lithium cacodylate pH 7.2 buffer containing 100 mM NaCl for at least one night at 4 °C. Further dilutions were made in the same buffer.FRET-melting assay
Denaturation of the oligonuceotide F21T (fluorescein-3′-GGGTTAGGGTTAGGGTTAGGG-5′-TAMRA) to probe the interaction of a ligand with G-quadruplex DNA was described elsewere.21a In the experiments presented here, a real-time PCR apparatus (Mx3000P, Stratagene) was used, allowing the simultaneous recording of 96 samples. Fluorescence measurements with the F21T oligonucleotide (0.2 µM) were studied in 10 mM lithium cacodylate pH 7.2 buffer containing 100 mM NaCl. The melting of the G-quadruplex was monitored alone or in the presence of 1 µM of compound, by measuring the fluorescence of fluorescein. To test the binding selectivity of the compound to the quadruplex structure, we added various concentrations of competitors: double-stranded DNA (self-complementary oligonucleotide ds26: 5′-GTTAGCCTAGCTTAAGCTAGGCTAAC-3′), tetramolecular G-quadruplex [TG5T]4 or intramolecular G-quadruplex (30AG: 5′-AGGGGGTTAGGGGGTTAGGGGGTTAGGGGG-3′). Emission of fluorescein was normalized between 0 and 1, and T1/2 was defined as the temperature for which the normalized emission is 0.5. T1/2 and ΔT1/2 are the mean of at least 2–4 values ± standard deviation.TRAP assay
The TRAP reaction was performed in a 20 mM Tris HCl pH 8.3 buffer containing 63 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 0.005% Tween 20, 0.1 mg ml−1 BSA, 50 µM dTTP, dGTP and dATP, 5 µM dCTP, and the oligonucleotides TS (5′-AATCCGTCGAGCAGAGTT-3′) (0.4 µM), ACX (5′-GCGCGGCTTACCCTTACCCTTACCCTAACC-3′) (0.4 µM), NT (5′-ATCGCTTCTCGGCCTTTT-3′) (0.4 µM) and TSNT (5′-ATTCCGTCGAGCAGAGTTAAAAGGCCGAGAAGCGAT-3′) (20 nM), 2 units of Taq polymerase, 0.02 mCi mL−1 of [α32P]-dCTP and 200 ng of A431 CHAPS extracts. After telomerase elongation for 15 minutes at 30 °C, 30 cycles of PCR were performed (94 °C, 30 s; 50 °C, 30 s; and 72 °C for 90 s). Telomerase extension products were then analysed on a denaturing 6% polyacrylamide, 7 M urea 1X Tris Borate EDTA (TBE) vertical gel. Extension products were quantitated using a Phosphorimager apparatus; telomerase relative activity was plotted against each compound concentration.Acknowledgements
M. K. thanks the European Union for a postdoctoral Marie Curie Intra-European Fellowship. This study has been supported by L'Association pour la Recherche contre le Cancer (grant #3482 to MPTF and #3365 to JLM) and an EU FP6 “MolCancerMed” grant to JLM. The help of E. Weyher-Stingl (MPI of Biochemistry, Martinsried, Germany) for the LC HR MS measurements is greatly acknowledged. David Monchaud (Laboratoire de Chimie des Interactions Moléculaires, College de France) is greatly acknowledged for his help with the graphics.References
- (a) S. A. Stewart, I. Ben-Porath, V. J. Carey, B. F. O'Connor, W. C. Hahn and R. A. Weinberg, Nat. Genet., 2003, 33, 492 CrossRef CAS; (b) J. Karlseder, A. Smogorzewska and T. de Lange, Science, 2002, 295, 2446 CrossRef CAS; (c) X. Zhang, V. Mar, W. Zhou, L. Harrington and M. O. Robinson, Genes Dev., 1999, 13, 2388 CrossRef CAS; (d) S. Neidle and D. E. Thurston, Nat. Rev. Cancer, 2005, 5, 285 CrossRef CAS.
- (a) Y. Mo, Y. Gan, S. Song, J. Johnston, X. Xiao, M. G. Wientjes and J. L.-S. Au, Cancer Res., 2003, 63, 579 CAS; (b) M. A. Shammas, R. J. S. Reis, C. Li, H. Koley, L. H. Hurley, K. C. Anderson and N. C. Munshi, Clin. Cancer Res., 2004, 10, 770 CrossRef CAS.
- (a) T. R. Cech, Cell, 2004, 116, 273 CrossRef CAS; (b) J. W. Shay, Cancer Res., 2005, 65, 3513 CrossRef CAS.
- (a) E. M. Rezler, D. J. Bearss and L. H. Hurley, Annu. Rev. Pharmacol. Toxicol., 2003, 43, 359 CrossRef CAS; (b) S. Neidle and G. Parkinson, Nat. Rev. Drug Discovery, 2002, 1, 383 CrossRef CAS.
- (a) S. Neidle and G. N. Parkinson, Curr. Opin. Struct. Biol., 2003, 13, 275 CrossRef CAS; (b) L. H. Hurley, Nat. Rev. Cancer, 2002, 2, 188 CrossRef CAS; (c) T. Simonsson, Biol. Chem., 2001, 382, 621 CAS; (d) R. H. Shafer and I. Smirnov, Biopolymers, 2001, 56, 209 CrossRef; (e) J.-L. Mergny, P. Mailliet, F. Lavelle, J.-F. Riou, A. Laoui and C. Hélène, Anti-Cancer Drug Des., 1999, 14, 327 CAS.
- (a) G. Pennarun, C. Granotier, L. R. Gauthier, D. Gomez, F. Hoffschir, E. Mandine, J.-F. Riou, J.-L. Mergny, P. Mailliet and F. D. Boussin, Oncogene, 2005, 24, 1 CrossRef; (b) D. Gomez, R. Paterski, T. Lemarteleur, K. Shin-ya, J.-L. Mergny and J.-F. Riou, J. Biol. Chem., 2004, 279, 41487 CrossRef CAS; (c) M. Sumi, T. Tauchi, G. Sashida, A. Nakajima, A. Gotoh, K. Shin-Ya, J. H. Ohyashiki and K. Ohyashiki, Int. J. Oncol., 2004, 24, 1481 CAS; (d) A. M. Burger, F. Dai, C. M. Schultes, A. P. Reszka, M. J. Moore, J. A. Double and S. Neidle, Cancer Res., 2005, 65, 1489 CrossRef CAS; (e) C. Leonetti, S. Amodei, C. D'Angelo, A. Rizzo, B. Benassi, A. Antonelli, R. Elli, M. F. G. Stevens, M. D'Incalci, G. Zupi and A. Biroccio, Mol. Pharmacol., 2004, 66, 1138 CrossRef CAS.
- (a) M. Read, R. J. Harrison, B. Romagnoli, F. A. Tanious, S. H. Gowan, A. P. Reska, W. D. Wison, L. R. Kelland and S. Neidle, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4844 CrossRef CAS; (b) P. J. Perry, A. P. Reska, A. A. Wood, M. A. Read, S. M. Gowan, H. S. Dosanjh, J. O. Trent, T. C. Jenkins, L. R. Kelland and S. Neidle, J. Med. Chem., 1998, 41, 4873 CrossRef CAS; (c) J.-L. Mergny, L. Lacroix, M.-P. Teulade-Fichou, C. Hounsou, L. Guittat, M. Hoarau, P. B. Arimondo, J.-P. Vigneron, J.-M. Lehn, J.-F. Riou, T. Garestier and C. Hélène, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3062 CrossRef CAS; (d) M.-P. Teulade-Fichou, C. Hounsou, L. Guittat, J.-L. Mergny, P. Alberti, C. Carrasco, C. Bailly, J.-M. Lehn and W. D. Wilson, Nucleosides, Nucleotides Nucleic Acids, 2003, 22, 1483 CrossRef CAS; (e) F. Keoppel, J.-F. Riou, A. Laoui, P. Mailliet, P. B. Arimondo, D. Labit, O. Petigenet, C. Hélène and J.-L. Mergny, Nucleic Acids Res., 2001, 29, 1087 CrossRef CAS; (f) R. A. Heald, C. Modi, J. C. Cookson, I. Hutchinson, C. A. Laughton, S. M. Gowan, L. R. Kelland and M. F. G. Stevens, J. Med. Chem., 2002, 45, 590 CrossRef CAS; (g) O. Y. Feodoroff, M. Salazar, H. Han, V. V. Chemeris, S. M. Kerwin and L. H. Hurley, Biochemistry, 1998, 37, 12367 CrossRef CAS.
- (a) M.-Y. Kim, H. Vankayalapati, K. Shin-ya, K. Wierzba and L. H. Hurley, J. Am. Chem. Soc., 2002, 124, 2098 CrossRef CAS; (b) M.-P. Teulade-Fichou, C. Carrasco, L. Guittat, C. Bailly, P. Alberti, J.-L. Mergny, A. David, J.-M. Lehn and W. D. Wilson, J. Am. Chem. Soc., 2003, 125, 4732 CrossRef CAS; (c) E. M. Rezler, J. Seenisamy, S. Bashyam, M.-Y. Kim, E. White, W. D. Wilson and L. H. Hurley, J. Am. Chem. Soc., 2005, 127, 9439 CrossRef CAS.
- (a) J.-F. Riou, L. Guittat, P. Mailliet, A. Laoui, E. Renou, O. Petitgenet, F. Mégnin-Chanet, C. Hélène and J.-L. Mergny, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2672 CrossRef CAS; (b) J. A. Shouten, S. Ladame, S. J. Mason, M. A. Cooper and S. Balasubramanian, J. Am. Chem. Soc., 2003, 125, 5594 CrossRef CAS; (c) P. M. Murphy, V. A. Philips, S. A. Jennings, N. C. Garbett, J. B. Chaires, T. C. Jenkins and R. T. Wheelhouse, Chem. Commun., 2003, 1160 RSC; (d) C. Granotier, G. Pennarun, L. Riou, F. Hoffschir, L. R. Gautier, A. De Cian, D. Gomez, E. Mandine, J.-F. Riou, J.-L. Mergny, P. Mailliet, B. Dutrillaux and F. D. Boussin, Nucleic Acids Res., 2005, 33, 4182 CrossRef CAS.
- (a) S. M. Haider, G. N. Parkinson and S. Neidle, J. Mol. Biol., 2003, 326, 117 CrossRef CAS; (b) E. Gavathiotis, R. A. Heald, M. F. G. Stevens and M. S. Searle, J. Mol. Biol., 2003, 334, 25 CrossRef CAS; (c) Q. Chen, I. Kuntz and R. H. Shafer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 2635 CrossRef CAS; (d) J. A. Schouten, S. Ladame, S. J. Mason, M. A. Cooper and S. Balasubramanian, J. Am. Chem. Soc., 2003, 125, 5594 CrossRef CAS; (e) I. M. Dixon, F. Lopez, J.-P. Estève, A. M. Tejera, M. A. Blasco, G. Pratviel and B. Meunier, ChemBioChem, 2005, 6, 123 CrossRef CAS; (f) A. T. Phan, V. Kuryavyi, H. Y. Gaw and D. Patel, Nat. Chem. Biol., 2005, 1, 167 CrossRef CAS; (g) M. A. Read and S. Neidle, Biochemistry, 2000, 39, 13422 CrossRef CAS; (h) R. J. Harrison, J. Cuesta, G. Chessari, M. A. Read, S. K. Basra, A. P. Reszka, J. Morrell, S. M. Gowan, C. M. Incles, F. A. Tanious, W. D. Wilson, L. R. Kelland and S. Neidle, J. Med. Chem., 2003, 46, 4463 CrossRef CAS.
- (a) Y. Wang and D. J. Patel, Structure, 1993, 1, 263 CrossRef CAS; (b) G. N. Parkinson, M. P. Lee and S. Neidle, Nature, 2002, 417, 876 CrossRef CAS; (c) L. Ying, J. J. Green, H. Li, D. Klenerman and S. Balasubramanian, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14629 CrossRef CAS; (d) S. Redon, S. Bombard, M.-A. Elizondo-Riojas and J. C. Chottard, Nucleic Acids Res., 2003, 31, 1605 CrossRef CAS; (e) A. T. Phan and D. J. Patel, J. Am. Chem. Soc., 2003, 125, 15021 CrossRef CAS; (f) J. Qi and R. H. Shafer, Nucleic Acids Res., 2005, 33, 3185 CrossRef CAS and references cited therein; (g) J. Li, J. J. Correia, L. Wang, J. O. Trent and J. B. Chaires, Nucleic Acids Res., 2005, 33, 4649 CrossRef CAS; (h) M. Vorlickova, J. Chladkova, I. Kejnovska, M. Fialova and J. Kypr, Nucleic Acids Res., 2005, 33, 5851 CrossRef CAS; (i) J. Qi and R. H. Shafer, Nucleic Acids Res., 2005, 33, 3185 CrossRef CAS; (j) D. Miyoshi, H. Karimata and N. Sugimoto, Angew. Chem., Int. Ed., 2005, 44, 3740 CrossRef CAS.
- (a) M.-P. Teulade-Fichou and J.-P. Vigneron, in Small molecule DNA and RNA binders: From Synthesis to Nucleic Acid Complexes, ed. M. Demeunynck, C. Bailly and W. D. Wilson, Wiley-VCH, Weinheim, 2003, vol. 1, ch. 11, pp. 278–311 and references cited therein Search PubMed; (b) J. Seenisamy, S. Bashyam, V. Gokhale, H. Vankayalapati, D. Sun, A. Siddiqui-Jain, N. Streiner, K. Shin-ya, E. White, W. D. Wilson and L. H. Hurley, J. Am. Chem. Soc., 2004, 127, 2944.
- F. Rosu, V. Gabelica, K. Shin-Ya and E. De Pauwn, Chem. Commun., 2003, 2702 RSC.
- (a) Y. Tor, ChemBioChem, 2003, 4, 998 CrossRef CAS; (b) F. Walter, Q. Vicens and E. Westhof, Curr. Opin. Chem. Biol., 1999, 3, 694 CrossRef CAS; (c) M. Zacharias, Curr. Med. Chem., 2003, 2, 161 CAS.
- J.-L. Mergny, et al., unpublished work.
- (a) D. P. Arya, R. L. Coffee, B. Willis and A. L. Abramovitch, J. Am. Chem. Soc., 2001, 123, 5385 CrossRef CAS; (b) D. P. Arya, L. Micovic, I. Charles, R. L. Coffee, B. Willis and L. Xue, J. Am. Chem. Soc., 2003, 13, 3733 CrossRef.
- (a) M. Hendrix, P. B. Alper, E. S. Priestley and C.-H. Wong, Angew. Chem., Int. Ed. Engl., 1997, 36, 95 CrossRef CAS; (b) D. Fourmy, M. I. Recht, S. C. Blanchard and J. D. Puglisi, Science, 1996, 274, 1367 CrossRef CAS.
- (a) S. M. Nelson, Pure Appl. Chem., 1980, 52, 2461 CrossRef CAS; (b) D. Chen and A. E. Martell, Tetrahedron, 1991, 47, 6895 CrossRef CAS; (c) J. Jazwinski, J.-M. Lehn, R. Méric, J.-P. Vigneron, M. Cesario, J. Guilhem and C. Pascard, Tetrahedron Lett., 1987, 28, 3489 CrossRef CAS.
- M. Sainlos, P. Belmont, J.-P. Vigneron, P. Lehn and J.-M. Lehn, Eur. J. Org. Chem., 2003, 2764 CrossRef CAS.
- J. L. Sessler and A. K. Burell, in Macrocycles, Topics in Current Chemistry, ed. E. Weber and F. Vögtle, Springer-Verlag, Berlin, 1992, vol. 161, p. 179 Search PubMed.
- (a) J.-L. Mergny and J.-C. Maurizot, ChemBioChem, 2001, 2, 124 CrossRef CAS; (b) F. Rosu, E. De Pauwn, L. Guittat, P. Alberti, L. Lacroix, P. Mailliet, J.-F. Riou and J.-L. Mergny, Biochemistry, 2003, 42, 10361 CrossRef CAS.
- (a) J. M. Sanders and C. Hunter, J. Am. Chem. Soc., 1990, 112, 5525 CrossRef CAS; (b) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210 CrossRef CAS.
- pKa's of the free neomycin are: 5.7, 7.6, 7.6, 8.1 (NH2 attached to the sugar rings); 8.6, 8.7 (NH2 attached to the two primary carbons).16a Thus, the free neomycin has a global charge of 4+–6+ depending on the experimental pH (7.2 for FRET-melting, 8.3 for TRAP). In the macrocyclic series, the two benzylic nitrogens have pKa's of around 7–8, and the ring nitrogens of the three aromatics are not protonated, neither are the two basic NH2 residues of neomycin engaged in the amide bound. Hence, the global charge of the macrocycles at experimental pH (7.2) is estimated to be around 4+ (amino caproic linker) and 6 + (lysine linker).
- P. Alberti, J. Ren, M.-P. Teulade-Fichou, L. Guittat, J.-F. Riou, J. B. Chaires, C. Hélène, J.-P. Vigneron, J.-M. Lehn and J.-L. Mergny, J. Biomol. Struct. Dyn., 2001, 19, 505 Search PubMed.
- J.-L. Mergny, A. De Cian, A. Ghelab, B. Saccà and L. Lacroix, Nucleic Acids Res., 2005, 33, 81 CrossRef CAS.
- J.-L. Mergny, J. Li, L. Lacroix, S. Amrane and J. B. Chaires, Nucleic Acids Res., 2005, 33, e138 CrossRef.
- D. Gomez, J.-L. Mergny and J.-F. Riou, Cancer Res., 2002, 62, 3365 CAS.
- A. J. Zaug, E. R. Podell and T. R. Cech, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10864 CrossRef CAS.
- (a) M.-P. Teulade-Fichou, J.-P. Vigneron and J.-M. Lehn, J. Supramol. Chem., 1995, 5, 139 Search PubMed; (b) O. Baudoin, M.-P. Teulade-Fichou, J.-P. Vigneron and J.-M. Lehn, J. Org. Chem., 1997, 62, 5454; (c) C. J. Chandler, L. W. Deady and J. A. Reiss, J. Heterocycl. Chem., 1981, 18, 599 Search PubMed.
- K. Wüthrich, NMR of Proteins and Nucleic Acids, John Wiley, New York, 1986 Search PubMed.
- A. Bax and D. G. Davis, J. Magn. Reson., 1985, 65, 355 CAS.
- J. Jeener, B. H. Meier, P. Bachman and R. R. Ernst, J. Chem. Phys., 1979, 71, 4546 CrossRef CAS.
- A. Bax and D. G. Davis, J. Magn. Reson., 1985, 63, 207 CAS.
- V. Sklenar, M. Piotto, R. Leppik and V. Saudek, J. Magn. Reson., 1993, 102, 241 CAS.
Footnote |
| † Current address: Chemical Genomics Centre, Otto-Hahn-Str. 15, 44227 Dortmund, Germany. |
| This journal is © The Royal Society of Chemistry 2006 |