Waste-free large-scale syntheses without auxiliaries for sustainable production omitting purifying workup
Gerd Kaupp
Faculty V, Organic Chemistry 1, University of Oldenburg, Germany, Diekweg 15, D-26188 Edewecht, Germany. E-mail: gerd.kaupp@uni-oldenburg.de; Fax: +49 4486 920704
First published on 25th September 2006
Abstract
Gas–solid and stoichiometric solid–solid reactions are usually quantitative and waste-free and thus particularly sustainable and environmentally benign. It is therefore shown how these can be scaled up with an emphasis for their use in chemical production. There is no necessity for purifying workup with solvent consumption (that means real solvent-freeness), waste disposal and other excessive energy input. These techniques avoid auxiliaries such as solvents, supports and catalysts, but the latter may be applied as gases with easy removal after reaction. Heat development is usually not a problem. Heat removal can be handled and is described also for extreme cases, as is the availability of equipment, in particular suitable milling techniques. The solid-state mechanism, which involves phase rebuilding (anisotropic molecular migrations within crystals), phase transformation and crystal disintegration, is the basics for the technical design. If (intermediate) melting cannot be avoided by cooling below eutectic temperatures, melt reactions at higher temperatures than required for solid-state reactions can be also rapid, uniform and quantitative, when direct crystallization of the product occurs. This version has an increased risk for side reactions as it does not profit from the crystal packing of the starting materials, but it profits from the crystal structure of the high melting product. For preparative purposes “large-scale” starts with the kg-range that can be easily reached in research institutes. It provides the basics for further scale up in industrial environments. Numerous typical examples are given for organic and some inorganic syntheses. These include solvent-free salt formations, complexations, condensations of amines, heterocyclic syntheses, Knoevenagel condensations, cascade reactions, halogen additions, stereo- and regio-specific protective reactions, and redox reactions. All of these may be of technical importance. Some of these involve now easily obtained products that cannot be achieved by solution reactions.
1. Introduction
Organic solid-state reactions are usually quantitative in a short time without the necessity of purification if excess gases can be easily removed or if stoichiometric solid–solid reactions are properly performed in mills. In the case of couple products such as H2O or gases, these are evaporated. If stoichiometric amounts of simple salts are the couple products and the organic material cannot be sublimed off, a minute amount of waste will occur by aqueous extraction of the salt, e.g. of NaCl, but these have highest possible atom economy and also belong to the most benign known processes. The reasons for the excellent performance and uniformity are favorable kinetics and the action of the supramolecular crystal packing with most regular self-assembled alignment of the reacting molecules and approach of the reagents in the crystal bulk that is profited from in the absence of intermediate melting. The solid-state mechanism has been elucidated by atomic force microscopy (AFM), scanning near-field optical microscopy (SNOM), and grazing incidence diffraction (GID) for very different situations and has been comprehensibly described.1 Preparative applications in more than 1000 waste-free reactions with 100% yield have been collected.2 The most important novelty are anisotropic far-reaching molecular migrations within the crystal along cleavage planes and channels that can also be achieved by mechanical interaction.3 It is now timely to discuss and collect the presently available larger scale production for technical applications, which was suggested in ref. 1 and 2. The techniques are easy to run and cheap as they save energy at lower temperatures than required in solution or melt and thus have many advantages over “solvent-free” reactions with microwave activation (these do not profit from the crystal structure bargain) that usually spend solvent for the distribution of the material to adsorbing supports prior to reaction and have to spend solvent again for the extraction of products from the spoiled support and for chromatography and recrystallization. The larger scale reactions reported here can, of course, be scaled up further in industrial environments. All of them provide directly pure products without the necessity of purifying workup, if the starting materials were pure and the stoichiometry was precise, so that no component was in excess. If gaseous catalysts are required in rare cases, these are removed by evaporation and if stoichiometric salts occur as by-products sublimation of the product or washings with water remove these.2. Gas–solid technique
Most gas–solid reactions are run close to room temperature and from ambient pressure down to low pressures at the end of reaction in tight previously evacuated vessels (static) or in diluted gas flows (dynamic). If excess gas is applied in a static technique the remainder is frozen out in a cold trap for further use. The same is true if another gas is formed upon reaction. In the latter case some mechanical agitation is necessary for efficient mixing of the gases during reaction. Static and dynamic systems are possible for large-scale reactions. Fig. 1 collects some equipment for what is possible in a research lab and further scale up is easily imagined. Round-bottomed flasks are the best choice for preliminary test runs on a smaller scale. These can be scaled up in desiccators or with closed rotovapor-type equipment with continuous or discontinuous supply of reacting gas and cooling/heating. The latter case provides mechanical agitation that can also be achieved by overhead rotation in closed flasks. These techniques are suitable up to the 500 g scale. Larger-scale and strongly exothermic gas–solid reactions require gas flow in stream bed or suspension bed and fluidized bed in columns with gas flow. Inert gas is admixed for cooling or heating, if necessary. The gas flow is adjusted for a sharp reaction front to occur. Melting has to be avoided, because reaction would stop in most cases. Therefore, external cooling to lower temperatures might be necessary.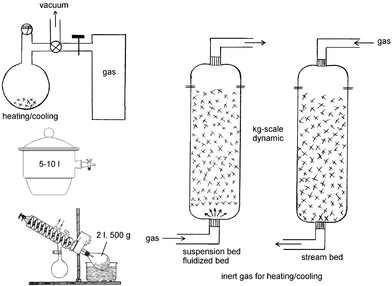 | ||
| Fig. 1 Equipment proposals for the execution of larger scale gas–solid reactions. | ||
3. Milling technique (solid–solid and rarely gas–solid)
Unlike gas–solid reactions, solid–solid reactions require continuous fresh contact between the reacting solids. This can be obtained by grinding or ultrasound irradiation, but milling is much more efficient and the only choice for larger-scale syntheses and production. For good and fast results it is essential to choose the least space demanding least noisy and most efficient milling technique with low energy consumption.Some of the equipment questions have already been addressed1 for gas–solid and solid–solid reactions in horizontal ball mills at sizes up to 2 l and this will not be repeated here. Industrial mills of different types are in practical use for a number of commercial applications. Among these, vibration mills have to move the whole mass of the grinding chamber. This produces high noise levels and environmental vibrations; it limits the sizes and the speed of the milling tools. Furthermore, they are almost impossible to be operated under controlled condition of atmosphere or closed circuit. The same is valid for simple (drum-) ball mills with a rotating vessel.4 Jet mills use large streams of air or inert gas, which limits their use in reactive milling and the absence of grinding media excludes significant kinetic effects. Horizontal or vertical bead mills do not exhibit a significant kinetic impact since no high-level relative acceleration of the grinding tools occurs. They experience shear- and friction effects but not collision.5 Planetary ball mills and shaker mills are limited to laboratory size. The most suitable choice is horizontal rotary ball mills with cooling/heating mantle that can be operated in dry milling at high relative velocity of the grinding media (up to 14 m s−1) that cannot be reached by the other technical ball mills. Furthermore, they run under controlled conditions like vacuum, inert gas or in closed circuits.6 The systems are presently available from 0.5 to 400 l grinding chamber capacity,7 and larger volumes seem to be possible. They are already in wide industrial use for mechanical alloying of different metals and/or ceramics and production of nanocrystalline metal hydrides,8–11 due to an intensive grinding effect at low energy costs (e.g. 600 W at 1200 rpm of the 2 l version) and short process times with lowest contamination of the processed powders by the milling tools, since the process is based on the collision of grinding media rather than on shear and friction.
Fig. 2 shows two cross-sections of the rotary mill. The side view (a) depicts the optimized shape of the rotor that drives the balls at up to 1800 rpm. The steel balls (Cr6 or ceramics) are pushed towards the walls while the rotor spins (frontal cross-section b) and the reacting crystals are milled between the balls and the wall. The materials are stellite and hard metal or ceramics (e.g. Si3N4 or ZrO2, etc), the latter for handling strongly corrosive reagents or products (e.g. the gases Cl2, HCl, HBr, etc) or in pharmaceutical and food industry. The ball diameters are 5 mm and the fill is 2 kg for a 2 l and 100 kg for a 100 l production unit.
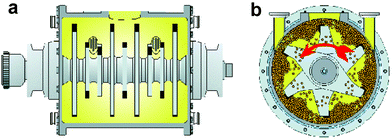 | ||
| Fig. 2 Cross-sections through the milling chamber with rotor; (a) side-wise; (b) frontal while rotating with balls; courtesy of Zoz GmbH, Wenden, Germany. | ||
The 2 l version of the horizontal rotary ball mill (Simoloyer® CM01-2l) with air-lock for loading operation and unloading under vacuum and/or inert-gas has been imaged in ref. 1. It can be easily run in a university lab, as the vibrational impact is very low. The industrial 100 l rotary ball mill in a plant environment has two synchronous drive motors and a stable base (Fig. 3). Again, the milling compartment is not moved and that makes it versatile for the industrial production. We address here the most efficient patented milling principle at highest relative speed of the milling balls (14 m s−1) that is only available in the Zoz Simoloyer® horizontal rotary ball mills and depict here the larger 100 l production unit, while 400 l mills of that type are also available.
 | ||
| Fig. 3 100 l production unit of a Simoloyer® CM 100-s2 ball mill of Zoz GmbH, set for inert gas operation; courtesy of Zoz GmbH, Wenden, Germany. | ||
The milling-out after reaction is performed with a semi-batch internal gas cycle charging and separation system with discharge through a cyclone, as already imaged in ref. 1. A complete description is found in ref. 12. The powders are collected by an internal air or inert gas cycle through a cyclone of the connected separation/classification system (Simoloyer® VS01a). The latter system is particularly advantageous because it allows for automatic reloading for the next batches prior to complete powder collection. When this setup is not available in an academic environment, milling-out through a grate insert that keeps the balls can be performed simply under gravity or more rapidly with a stream of gas that may be enhanced by a vacuum cleaner with collection through a cyclone avoiding losses. Holdup is typically 10% but this counts only for the last batch. The preceding batches give the 100% yield of product with the same purity as the starting materials. Inert gas is used for sensitive solids preferably in the internal gas cycle version.
4. Experimental
Caution:Gas–solid reactions may be highly vigorous if finely ground crystals are applied. Do not apply preceding or supporting milling unless the reaction is incomplete due to surface passivation.Caution:Do not mill explosive solids, as these might explode upon the heavy shocks despite the presence of the steel balls with high heat capacity.
Caution:Do not mill under enclosed air but under vacuum or inert gas if hydrogen or methane or other burning gas is a reaction product in order to avoid explosive gas mixtures.
Caution:Do not use milling for oxidations of highly reducing solid materials with oxidizing gases such as nitrogen dioxide or oxygen that might become violent with micronized dusts.
It is essential that the equipment be gas-tight. Gas–solid reactions in closed systems are usually performed with excess gas for complete reaction, the excess being recovered in cold traps. It is, however also possible to remove traces of exhaust gases down to the detection limit with excess solid and complete the chemical transformation of the solid in a second step with a suitable amount of the same gas. The gases are usually fed-in from pressure bottles. If vapors of low boiling liquids are applied a slightly higher temperature in the reaction vessel at the site of the crystals as compared to the connected vessel with the vaporizing liquid at a vacuum line must avoid gas condensation. More secure is the connection of the evacuated reaction vessel to a sufficiently large container with fully vaporized liquid at a vacuum line. The ratio of reacting and inert gas and the flow rate in the dynamic mode have to be carefully evaluated for any new reaction. It might be essential to increase the concentration of reacting gas towards the end of reaction in those cases.
Starting crystals should be >99% pure if pure products are aimed at without workup, as any minor impurities will persist in the product. However there may also be cases where impurities can be more easily removed (by extraction) from the quantitatively obtained product than from the starting material.13 In that case the stoichiometry must take into account the percentage of the minor impurity for avoiding excess of one reagent in the case of solid–solid reactions.
General procedure for solid–solid reaction in the 2 l mill that can be scaled for the range of 0.5 to 400 l for nearly thermoneutral production:
200 g quantities of stoichiometric mixtures of the loosely premixed crystalline reaction partners were fed to the 2 l stellite ball mill with hard metal rotor and 2 kg of 5 mm steel balls (Cr6) and tape-water cooling. The rotor speeds were 800–1300 min−1 (a maximum of 1800 min−1 is available). The time was mostly <30 min for complete conversion, the temperature at tape-water cooling was 14 °C at the walls and 20–30 °C in the center of the mill. Without cooling a maximum temperature of 40–60 °C was reached during milling for 30 min if the reaction was close to thermoneutral. Overall, a ΔG-value of <−6 kcal mol−1 is required for principally reversible quantitative overall processes including the contributions of the crystal energies. The product crystallites were usually sub-micrometer in diameter, but these often aggregated to clusters. The completeness of reaction was checked by suitable analytical techniques (IR, NMR, MS, DSC, etc). Milling-out by gravity or in a stream of air or inert gas for about 10 min was performed with varying rotor speeds for best results (600, 900, 1200 min−1). The next batch was loaded and the sequence repeated, etc. The yield per batch of pure product was 100% by weight on the average.
Variant with gas formation
If a gas is formed upon solid–solid reaction as in the case of salt formations with alkali (hydrogen)carbonates it is essential to control the internal pressure that evolves during milling. For example, in a typically one molar reaction in the 2 l mill, the pressure would roughly approach 15 bar if it were not released by a pressure valve with protection by a dust filter. The gas can be collected if it is a desired product or used for the milling-out cycle that follows after gas production has ceased.Variant with reactant gas
Only gas–solid reactions with surface passivation (if phase transformation or crystal disintegration is too slow) require milling for completion. No milling should be applied in the absence of surface passivation because the reaction would become violent. The reacting gas might be continuously added through a valve under slight pressure of about 3 bar.In the reaction with NaNO2 the reacting gas NO2 (in equilibrium with N2O4) was transformed to the product gas NO at 0.8 bar (ca. 0.07 mol in the 2 l mill with balls and reagent) and completely transformed by 30 min milling with an initial excess of NaNO2 (200 g), prior to collection by filling to a pressure bottle, prior to the next batch with NO2, etc until the reagent was completely transformed to NaNO3.
Analyses
The completeness of reaction and purity of the products were checked by weight increase, gas pressure (gas–solid reactions), melting point, solid-state IR and mass spectroscopy, followed by titration, thin layer chromatography, 1H-NMR, or other more specific analysis techniques. It is important that the solid-state techniques be performed first. For example, if the IR test indicated completion one continued with the milling for another 20% of the reaction time. The samples were taken from the surface, center, and bottom of the crystal masses in solid–gas reactions unless these were uniform due to efficient agitation during reaction. Sampling in solid–solid reactions was at the beginning and the end of the milling-out. Homogeneous crystal form and size were secured by light microscopy at 400-fold magnification.5. Large scale reactions, general remarks
From the more than 1000 quantitative waste-free solid-state reactions that are known since 19851,2 we chose some for scale up with low-priced solid reagents for varied important situations in order to assess the versatility. For example, there may be the addition type (e.g. complexation), or there may be condensation with formation of the couple product water, which is included into the product crystals as crystal water. If the water of reaction cannot be accommodated, a drying agent (for example MgSO4·2 H2O) would be necessary to remove liquid water. Other situations occur if alkali salts are by-products or if a gas is evolved as a couple-product, and finally one can perform sluggish gas–solid reactions with milling support as in the production of reactant gases, if surface passivation must be removed by continuously breaking the reacted surface region.New solid-state reactions should be planned according to sections 1, 2, and 3. Furthermore, the presently reported examples and those in ref. 1 and 2 can serve as important guidelines. Ceramics mills and tools are available for corrosive situations. Melting must be avoided if the benefits of the crystal-packing bargain shall be used (melt reactions require considerably higher temperatures and run increased risk of side reactions). Therefore, cooling mantles (also usable for heating) must be provided at the mills and cooling below room temperature depends on the eutectic temperatures. If the melting points/eutectic temperatures are very low (e.g. benzaldehyde 16 and aniline 17), stoichiometric melt reactions without catalysts can be more profitable than cooling below −20 or −30 °C for solid-state production, in particular when no side reactions occur and if the product crystallizes directly at the higher reaction temperature. The worst choice would be dilution with solvents and addition of catalysts. While the heat capacity of mill, rotor, and balls is very high and many solid–solid reactions are almost thermoneutral, cooling is required for exothermic reactions to remove the heat. Excessively exothermic solid–solid reactions should be avoided. For example, solid acid and solid NaHCO3 should replace salt formations between solid acid and solid NaOH, etc. In solid–gas reactions extremely exothermic reactions (e.g. 1 bar CO2 and sodium alkoxides that would char) can be moderated and handled on a large scale by dilution of reacting gas and a flow of inert gas for heat removal (see Fig. 1 and 4). At moderately exothermic gas–solid reactions, larger crystals and constant feed of gas at lower pressure with external cooling bath are the method of choice (see Fig. 1 and e.g.31 or 36).
 | ||
| Fig. 4 Highly exothermic gas–solid reactions that require moderation by admixture of inert gas in flow systems. | ||
Every reactive crystal that accepts reagents for reaction by necessity exhibits cleavage planes or channels of sufficient size for enabling the molecular migrations of reagents and products in the phase rebuilding step. Surface passivation due to difficulties with phase transformation and disintegration of the phases is rarely observed with supramolecular crystals.1,2 These instances are automatically engineered to become successful by the milling for the solid–solid reactions. At gas–solid reactions milling should only be applied if passivation of the surfaces has been secured. It might be necessary to do it cautiously (for example, at low rate and low gas pressure) in order to avoid violent reaction.
The ingenious three-step phase-rebuilding solid-state mechanism avoids decrease of reactivity by solvation (as it occurs in solutions or melts). Therefore catalysis or activating substitution of the reagents is mostly unnecessary. It would be at the expense of atom economy and suitable melting points. For example, solid-state ester syntheses occur between the comparatively high melting acids and solid alcohols (for example oxalic acid and cholesterol, slightly heated in the 1 ∶ 2 ratio esterifies waste-free). It is not necessary to use esters (usually lower melting) or other activated acid derivatives or activated alcohol derivatives (all introducing wastes that are subject to removal) with the risk to end with liquids due to the spoiling liberated leaving groups. Other examples are amino acids (e.g.22 or cysteine, etc.1,2) that can be directly used in numerous solid-state reactions1,2 instead of activated amino acids, etc. The water of reaction here and in condensation reactions is often included as crystal water of the product and can be removed by evaporation in a vacuum. The addition of easily removable drying agents (for example MgSO4·2 H2O) is only necessary if the water of reaction separates from the crystals. Further favorable secondary products are gases that escape and may be collected as useful reagents. The choice of high melting starting materials is therefore also a choice of highest atom economy.
6. Salt formation
Unlike solution reactions, there are no problems for generating pure hygroscopic salts of weak bases with gaseous HCl or HBr. This has been checked with unground crystals of the amino acids (L)-phenylalanine, (D)-penicillamine (1), (DL)-penicillamine, (L)-cysteine, (L)-leucine, (L)-proline, (DL)-tyrosine and others at the 100 g scale in the closed rotovapor setup (Fig. 1) using a water bath or an ice bath. The pure hydrohalides were obtained free of water. The bis-hydrochloride was obtained with (L)-histidine (3) and HCl gas. Some of the amino acids, in particular glycine and alanine exhibited surface passivation if previously exposed to moist air. In these cases anhydrous premilling enabled the slightly exothermic complete salt formation.2 Preparative applications are also reported for the 100 g scale preparation of the dihydrochlorides of o-phenylenediamine (5) with the rotovapor equipment.14 The reactive gas was continuously let in from a steel cylinder by using the control needle valve for maintaining a pressure of 0.5 bar and by immersing the rotating flask into an ice bath. After about 1 h, when the gas uptake ceased, the HCl pressure was increased to 1 bar, the ice bath removed and the closed system left overnight. Excessive HCl was frozen out in a cold trap for further use and the pure solid dihydrochloride 6 (acid/base titration) stored at a dry site. If the monohydrochloride is required, the dihydrochloride (6) is milled with a stoichiometric amount of o-phenylenediamine (5) for 30 min. Numerous further weak bases’ hydrohalogenides (e.g. from benzotriazoles, benzothiazoles) and extremely sensitive hydrohalogenides of Schiff bases have been prepared easily and in good shape by gas–solid reactions2 but not all of them yet at large scale, which, however, should succeed if required.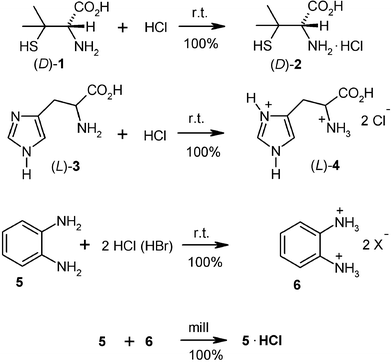
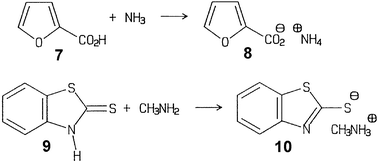
Equally important are hygroscopic ammonium salts of weak acids that can hardly be prepared in pure form from solutions. Again, exclusion of water is not a problem at all for gas–solid chemistry. This includes virtually all of the solid carboxylic acids but also phenols, imides and even 1,3-diketones (e.g. barbituric acid). The techniques are the same as in the reactions with HCl/HBr, and there is no reason why these should not be performed on a large scale.2 For example, the reactions of furan-2-carboxylic acid (7) with ammonia and of 2-mercaptobenzothiazole (9) with methylamine have been performed at the 100 g scale with the rotovapor equipment and proper cooling.15
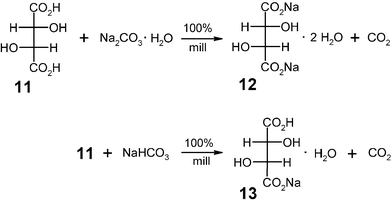
The solid–solid reactions of carboxylic acids and alkali hydroxides or alkali oxides are too violent for practical purposes. Therefore, the cheaper alkali (hydrogen)carbonates are the proper choice. The possibilities are manifold and it is avoided to dissolve the reactants in water, control the heat of neutralization and evaporate all of the water with very high energy consumption. If a dibasic acid such as (L)-(+)-tartaric acid (11) is needed as a food additive, either as the mono- (13) or disodium salt (12), it is only necessary to use either sodium hydrogencarbonate or sodium carbonate (for practical reasons as the solid monohydrate). These reactions have been performed as semi-continuous 200 g batches with quantitative yield and atmospheric air was the gas for the product collection from the mill.16 The solids are ready for use. Their dehydration will rarely be necessary.
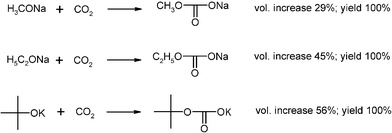
Extremely exothermic acid–base reactions with 250 g quantities of alkali alcoholates and carbon dioxide (Fig. 4) have been moderated by inert gas admixture in the suspension bed technique (Fig. 1).17,18
The highly versatile and merchandisable carbonic acid half-ester salts cannot be synthesized in pure form from solution. The CO2 concentration had to be gradually increased in N2 inert gas and there was a sharp reaction front that was kept at temperatures below 50 °C (charring occurs in a static system). The powder volume increased considerably (Fig. 4), so that one-third or one-half of the column had to be provisioned for that. At the end of the reaction pure CO2 was applied and the closed column left overnight. The products titrated correctly, as the alcohol impurities in the starting materials were expelled during the reaction. A very detailed experimental description is given in ref. 2.
7. Complexations
Quantitative complexations by stoichiometric milling of the partners are now common2 and many new molecular ratios of the components can be achieved in the solid state that are not available from solutions. Melting point, solid-state IR, CD and X-ray powder analysis are very useful techniques for the analysis. Practical applications are in (chiral) inclusion, charge transfer, and magneto chemistry.2,19 Due to only minor thermal effects there do not seem to exist limitations for the scaling up. An example is the large-scale 1 ∶ 1 complex formation of glucose (14) and urea (15). Previously, only mg quantities have been obtained by tedious and wasteful crystallization that took six months (until proper seed was obtained).20 By 5 min milling in the 2 l horizontal ball mill 200 g batches of the complex are obtained.1,2,16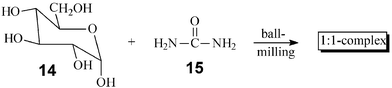
8. Condensations of amines
Many solid primary and secondary amines are well suited for condensation reactions with carbonyl compounds. This can be used for removal of the latter from exhaust gas down to the detection limit and for syntheses of imines or N/O-acetals or azomethine ylides or azomethine imines. Numerous waste-free and catalyst-free solid-state syntheses of these types have been collected in ref. 2 and there is no doubt that most of these can be scaled up. We describe here some that have been performed on a larger scale.A careful selection of “acidity” makes it possible to quantitatively remove acetone below the detection limit from exhaust gases in flow rates that mimic industrial flows. The reagent is hydroxylammonium phosphate in stoichiometric mixture with K2HPO4·xH2O so that free acetone oxime (CH3)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NOH is directly expelled from a two-column system at 80 °C together with the water, and the value added analytically pure KH2PO4 is the second useful product (1). A stream-bed approach with heated columns is applied here. The reacted column was contracted by less than 10%.
NOH is directly expelled from a two-column system at 80 °C together with the water, and the value added analytically pure KH2PO4 is the second useful product (1). A stream-bed approach with heated columns is applied here. The reacted column was contracted by less than 10%.
That procedure is also the easiest way to synthesize pure free acetone oxime when higher loads of acetone vapor might be applied. The acetone oxime was separated from water by continuous azeotropic removal with t-butyl methyl ether.14,21
| (H2NOH)3·H3PO4
+ K2HPO4·xH2O + 3 Me2CO → 3 Me2CNOH + 2 KH2PO4
+
(x
+ 3) H2O | (1) |
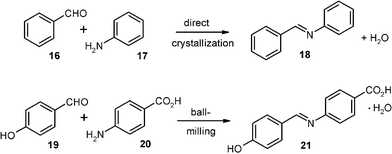
Schiff bases are important intermediates, the liquid-state synthesis of which is usually performed with wasteful acid catalysis and exclusion/removal of water due to easy hydrolysis. If starting materials such as aniline (17) and benzaldehyde (16) have very low melting points their condensation cannot be run as solid-state reaction. It has thus been performed by 900 W microwave activation of 106 + 93 mg of the reagents after adsorption to 20 mg of montmorillonite K-10 clay at 110 °C and final extraction from the support with CH2Cl2.22 Unfortunately, this process became a “highlight of green chemistry” as a so-called “solvent-free” reaction.23 However, such an effort constitutes extreme waste production use solvent and energy misuse.
Another favorable solid-state effect can be used for turning this synthesis to a sustainable and benign production technique: the direct crystallization of the product (18) out of the stoichiometric undiluted liquid mixture of the reagents 16 and 17 at the reaction temperature. Thus, if 848 g of 99.5% technical aniline (17) and 744 g of 99.5% technical benzaldehyde (16) are mixed at 18 °C in a flat steel pan (31 × 44 cm2) a slightly exothermic reaction ensues with slightly exothermic crystallization (maximal temperatures were 32 and 35 °C) within 12 min. On the next day, the wet crystals were crunched with an ordinary household grain-mill and dried in a vacuum at room temperature to give 1.438 kg of pure benzylidene–aniline (18).2,24 This simple energy saving technique so extremely supersedes all waste producing previous techniques in all respects that it must be shown here: it also profits from crystal effects providing a sufficiently high melting point of 18. There are no problems with hydrolysis equilibrium in a slightly exothermic reaction directly providing the product crystals out of the liquid. Any very minor quantities of 16 and 17 that might still be dissolved in the separated water of reaction (126 g) had also formed the crystalline 18 upon the evaporative drying. The absence of an acid catalyst is very important for the resistance of the Schiff base 18 towards hydrolysis.
Solid derivatives of aniline and benzaldehyde work equally well by stoichiometric milling also with 200 g batches provided that the water of reaction is taken up as crystal water by the product.2 An example is the reaction of 19 and 20 giving pure 21 by 15 min milling without cooling at up to 50 °C.2 The crystal water can be removed at 80 °C in a vacuum without loss by hydrolysis.16,20
Similarly, there can not be reasonable doubts that large-scale solid-state syntheses of quinoxalines (some heating is required, but melt reactions require considerably higher temperatures) from o-phenylenediamines and benzils2 or enamine ketones from solid 1,3-diketones and solid aniline derivatives2 would also succeed in 200 g batches and beyond, as the crystals stay dry upon stoichiometric milling at room temperature. If the product crystals cannot take up the water of reaction in these and related condensation reactions, neat reaction is obtained by addition of an easily removable solid drying agent such as MgSO4·2H2O. Alternatively, the stoichiometric components can be separately micronized by milling, the dusts mixed and heated to the reaction temperature in vacuo for continuous removal of the water as it forms.2 Unlike solution reactions with acid catalysis, there are no problems with hydrolysis.
Solid secondary amines and carbonyl compounds (or their hydrates) give enamines, N/O-acetals, or azomethine ylides and elusive azomethine imines. 200 g batches have been successfully performed by stoichiometric milling of (L)-proline (22) with paraformaldehyde to give the stable N/O-acetal 23, which assumes the zwitterionic form by reprotonation and takes up the water of reaction (no hydrolysis in the absence of acid catalysts).2 The corresponding solid-state hydroxymethylation occurs also with imidazole (0 °C) and benzimidazole (r.t.). These reactions involve initial monomerization of paraformaldehyde to give formaldehyde by polymer chain breakage. Suitable crystal packings enable the inserting additions of formaldehyde by providing channels (22 and benzimidazole) or cleavage planes (imidazole) that permit the necessary molecular migrations within the crystal upon reaction.
A stoichiometric 200 g run of 22 and ninhydrin (24) gave quantitative three-cascade solid-state reaction to provide the pure azomethine ylide 25 in a large quantity (146 g, 100%) after 40 min milling without difficulties, even though the crystals did not accept all of the water of reaction. The complete collection of the product was achieved by four times milling-out of the holdup portion with 250 ml water each and collection of this part of the yield by centrifugation and drying.16 Clearly, the addition of a drying agent (MgSO4·2H2O) would have enabled the semicontinuous multi-batch processing followed by easy washing-out of the drying agent.
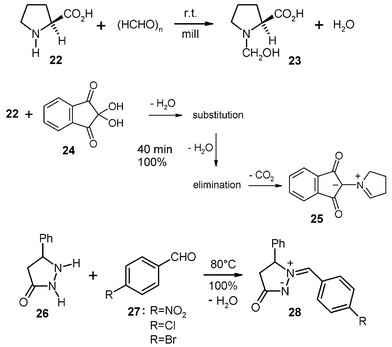
The pyrazolidinone 26 is a cyclic hydrazide that can loosen the H from the nitrogen next to the carbonyl group for water elimination after initial addition of the carbonyl group of 27. Thus, several stable azomethine imines 28 have been obtained by stoichiometric milling with solid benzaldehydes 27.2 Milling of the nitro-derivative 27 and 26 gave a quantitative yield of 28 (R = NO2) in a stoichiometric 200 g run. Thus, azomethine imines are now easily available on a large scale and there can be no doubt that 28 (R = Cl and Br) would also be quantitatively obtained on a larger scale and there is no hydrolysis due to the absence of an acid catalyst also upon removal of the crystal water from 28 at 80 °C in vacuum.
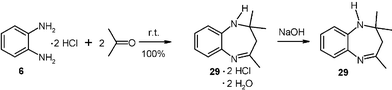
It is also possible to perform gas–solid amine condensations with vapors of liquid acetone or liquid amines. Thus, moderately large scale (20 g) cyclizing condensations have been performed with acetone vapor to the bis-hydrochloride 6, (DL)-penicillamine hydrochloride and (L)-cysteine hydrochloride.14 For example, the dihydro-1,5-benzodiazepine dihydrochloride 29·2 HCl hydrate and there from the free base 29 were quantitatively obtained if 20 g of o-phenylenediamine dihydrochloride crystals (6) was placed in an evacuated 10 l desiccator and connected to a 100 ml flask with 11.6 g degassed frozen acetone. After removal of the cooling bath the acetone evaporated smoothly, and the system was left overnight. The excess acetone was condensed to a cold trap and the dihydrochloride dihydrate of 29 quantitatively obtained. It can be liberated to the free base 29 with aqueous NaOH.
Similarly, the thiazolidine hydrochlorides can be obtained from the above mentioned aminoacid salts. The latter two reactions cannot be performed with the free amino acids due to surface passivation that has been found by AFM studies. However, the milling of (L)-cysteine with paraformaldehyde yields quantitatively (R)-thiazolidine-4-carboxylic acid, as the milling breaks the passivation.25 The easy gas–solid synthesis of benzylidene alkylimides by reaction of gaseous methylamine or ethylamine with solid benzaldehydes25 is still awaiting its scale up.
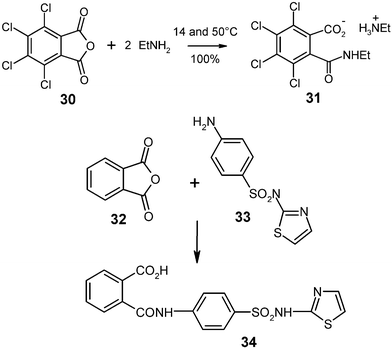
Ring opening amine condensation with gaseous amines has been obtained with 500 g quantities of tetrachlorophthalic anhydride (30)
(mp 255 °C) and ethylamine. The ethylammonium salt 31 was quantitatively formed in a 2 L flask at the rotovapor equipment (Fig. 1) and cooling with a water bath of 14 °C. The ethylamine was continuously let in by adjusting the pressure to 0.5–0.8 bar with a needle valve. Due to aggregation of 40 µm particles that formed grains of 0.5–3 mm diameter the reaction had to be interrupted at 75% conversion. After grinding, the continued reaction was completed at 50 °C for a quantitative yield.26 Unsubstituted solid anhydrides were less suitable for this type of large-scale reaction due to high heat production, but the known phthalimide type ring openings with gaseous amines show promise for successful scale up. Furthermore, solid aromatic amines such as 33 do not produce much heat upon reaction with phthalic anhydride (32) and can be stoichiometrically milled in 200 g batches with quantitative yield at room temperature for 1 h.15 The product 34 is free from bisamide side products, which occur upon reaction in solution or in the melt. The solid-state synthesis of this antibacterial sulfonamide phthalazole (34) was first described in ref. 27.
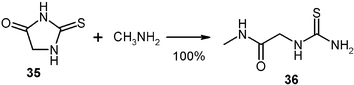
Numerous solid thiohydantoins were quantitatively ring opened with gaseous amines to give thioureido-acetamides (36)28 that are versatile building blocks in organic synthesis.29 These can be obtained in larger quantities, for example by reacting 100 g of thiohydantoin (35)
(mp 231 °C) with methylamine in a 2 l flask at the rotovapor equipment (Fig. 1) with continuous addition of the gas at 0.8 bar and cooling with a water bath of 14 °C. After the gas uptake ceased, the setup was filled to 1 bar of the reacting gas and left overnight for a quantitative yield (127 g) of 36 after recovery of the excessive gas to a cold trap.15 The specific cleavage site is between N and CO.
9. Heterocyclic syntheses
Phenacylbromide30 or chloroacetyl substituted heterocyclics31 have been used for the waste-free solid-state 3-cascade synthesis of varied 2-aminothiazoles, which are versatile building blocks in heterocyclic chemistry. It is therefore essential to scale up the procedure in large ball mills, as the technique of complete stoichiometric reaction removes the difficulties with lachrymatory reagents. The variations (e.g.39, 39a, 41, 42) are manifold. As there is no risk for melting and as not much heat is involved, all should be ready for scale up. Thus, 200 g batches of the basic reaction with stoichiometric mixtures of 37 and 38 have been successfully and quantitatively performed.16 The product salt 39 had no residual lachrymatory properties. Washings with aqueous NaOH can easily liberate the free base of 39, if required.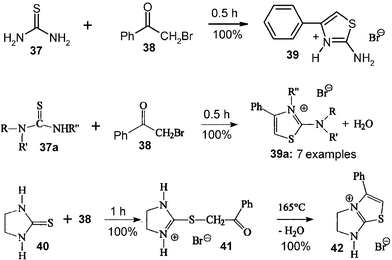
10. Knoevenagel condensations
Various waste-free C–C forming solid–solid reactions have been performed and some of these already scaled up. Typical solid C–H active compounds are dimedone, Meldrum's acid and barbituric acids that all condense in the solid state without catalysts.32 The products are interesting functional alkenes that can now be easily obtained in large quantities without the requirement for purifying workup. For example, stoichiometric millings with 200 g batches were successfully performed with p-hydroxybenzaldehyde (43) and barbituric acid (44a) as well as N,N′-dimethylbarbituric acid (44b) to give quantitative yields of 45a/45b.32 These compounds can be used in follow-up syntheses. The first reaction requires slightly higher temperatures that are achieved in the mill at 1200 rpm rate without cooling. The polar product crystals take up the water of reaction. It can be evaporated at 80 °C in vacuum, if required. Dimedone and benzaldehydes undergo Knoevenagel condensations followed by Michael addition that are quantitative with 1 ∶ 2 solid mixtures at 50–100 °C forming stable symmetric bis-enolketones by passing an intermediate melt with direct product crystallization (not shown here).32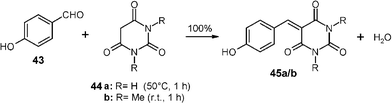
11. Further waste-free organic solid-state reactions
Almost all of the more than 1000 waste-free organic gas–solid and solid–solid reactions have the potential for being scaled up. The conditions are summarized in section 5 and countless further syntheses according to the methodologies described here, are firmly envisioned.33 The reported examples concentrate around low-prized or easily synthesized starting materials. It appears valuable to list here four syntheses of particular interest even though the latter three have not yet been scaled up, but there can be no doubt that their scale up will also be successful. These are addition of bromine to cholesterol (46),34 tetrabromination of tetraphenylethene (48),35 and the protection of the natural polyols (D)-mannitol (50) and myo-inositol (53) with phenylboronic acid (51)36 that all run waste-free with quantitative yield and full regio- or stereo-specificity, unlike solution reactions. They occur because the reagent's crystal packing involves either suitable cleavage planes or channels as in all other successful solid–gas and solid–solid reactions.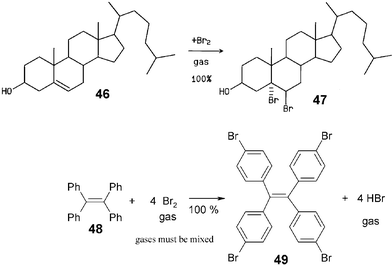
The industrially important enantiopure cholesterol dibromide 47 from solid–gas synthesis does not require purifying workup and has better properties than the wastefully produced 47 from acetic acid solution. The remarkable specific tetrabromination of 48 provides 49 as a useful core for dendrimers. Compound 49 is quantitatively obtained in pure form. The gases have to be efficiently mixed by rotation of the reaction vessel during reaction. Hydrobromic acid is a useful couple product for further use after separation from a slight excess of bromine by vacuum distillation while thawing the cold trap from −196 to −78 °C.
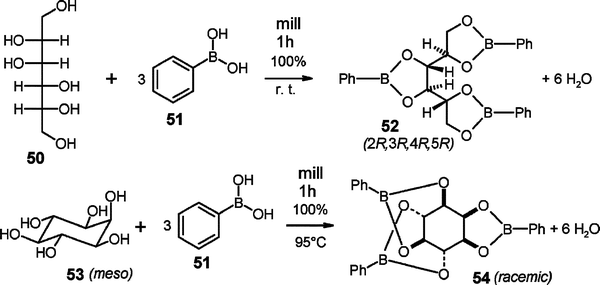
The stereo- and regiospecificity in the waste-free syntheses of (2R,3R,4R,5R)-52 or rac-54 by stoichiometric milling of 50 or 53 with 51 at room temperature or 95 °C, respectively,36 are remarkable. The reacting crystals exhibit proper channels for the necessary molecular migrations.
12. Some inorganic solid-state syntheses
If infinitely covalent crystals (e.g. SiO2, Al2O3, Si, WC, etc), which give plasma reactions (tribochemistry with multiple cleavages of sigma bonds upon cleavage of the crystals that belongs to mechanochemistry) upon milling, are excluded, there is no reason why inorganic crystals should not follow the same rules in quantitative solid-state syntheses, as do organic crystals. Such reactions are quantitative with molecular crystals (e.g. S8), salts and metals, as well as with infinite monolayered oxides (e.g. PbO), despite their sometimes very high melting points.An interesting production of the valuable gas nitrogen monoxide uses the reaction of nitrogen dioxide with sodium nitrite crystals (layered structure) (2). This reaction requires milling as the crystal surface passivates itself in this process.
| NO2 + NaNO2 → NO + NaNO3 | (2) |
The synthesis is performed with 200 g batches of NaNO2 in the 2 l horizontal ball mill by filling with 0.8 bar of the brown NO2 gas (0.07 mol) and collecting the colorless NO gas in a pressure bottle after 30 min milling. The reaction is continued with fresh NO2 until all NaNO2 has been transformed, etc.16
Complex salts can be easily produced by stoichiometric milling and also safely with highly poisonous powders in 200 g batches. This has been verified with copper cyanide (νCN = 2164 cm−1) and sodium cyanide (νCN = 2091 cm−1) in quantitative reactions for the production of the complex salts that are important in galvanotechniques. Depending on the molar ratio the different complex salts in (3), (4), and (5) are obtained.16
| CuCN + NaCN → Na [Cu(CN)2] (νCN = 2112 cm−1) | (3) |
| CuCN + 2 NaCN → Na2 [Cu(CN)3] (νCN = 2109 cm−1) | (4) |
| CuCN + 3 NaCN → Na3 [Cu(CN)4] (νCN = 2080 cm−1) | (5) |
Elementary sulfur is very corrosive to metals when ball milled. Even the steel milling materials are attacked and therefore ceramic mills should be used, but the stellite housing and hard-metal rotor were sufficiently resistant when ZrO2 balls were used. 200 g stoichiometric batches have been successfully performed with copper (6), (7) and iron (8) powders.16 These were chosen due to the importance of Cu2S as a useful solid for the production of luminophores and thermoelements, whereas FeS is used for the generation of H2S. Interestingly, changing the molar ratio of copper and sulfur allows also the clean synthesis of CuS.
| 16 Cu + S8 → 8 Cu2S | (6) |
| 8 Cu + S8 → 8 CuS | (7) |
| 8 Fe + S8 → 8 FeS | (8) |
The S8-reactions require mechanochemical S–S bond breakages by the milling prior to the chemical reaction. These waste-free low-temperature syntheses may be useful and many applications of sulfur and pyrite for the synthesis of metal sulfides might be envisaged.
Also high melting oxides such as lead oxide (orthorhombic, layered structure; mp 888 °C after phase transition at 490 °C to layered tetragonal structure) and chromium trioxide (mp 197 °C) combine to Chrome yellow (lead chromate, salt, mp 844 °C) (9), which can be further transformed to Chrome red with additional lead oxide by stoichiometric milling at room temperature (10). Some mixed oxides are important as ferroelectrica (PLZT ceramics) with high dielectric constants for optoelectronic and piezoelectric applications. Surprisingly, the high kinetic stoichiometric reactive milling (14 m s−1) also produced lead titanate (mp 450 °C) in 400 g batches with PbO (mp 888 °C) and titanium dioxide (rutile or anatase, mp 1830–1850 °C) (11). The syntheses with TiO2 must rely on tribochemical reactions, as metal–O covalent bonds must be broken from the infinite 3-D structures (exhibiting channels in rutile and anatase). Numerous further applications await their exploration (e.g. PtO2 + H2 in the presence of channels, etc).
| PbO + CrO3 → PbCrO4 | (9) |
| PbCrO4 + PbO → PbO·PbCrO4 | (10) |
| PbO + TiO2 → PbTiO3 | (11) |
13. Conclusions
The scale up of numerous organic and some inorganic solid-state reactions of various types without solvent and giving quantitative yield has been shown in the kg-range. The selection out of more than 1000 successful g-scale gas–solid and solid–solid reactions was largely governed by the prize of the starting materials and by the widespread use of the products obtained. There is no better way to save energy and time and to avoid waste than by these well-founded techniques and further scale up to the industrial range is clearly indicated. Technical solutions with strongly exothermic and with close to thermoneutral procedures are demonstrated for various circumstances. The first requirement for solid-state reactions is that there are no liquid phases, as this will remove the advantages of using favorable crystal structure effects as a bargain for not dissolving or melting. Melt reactions also profit from high concentration, but the reaction temperatures are considerably higher in the absence of the favorable crystal effects and they often bear the risk of side reactions. In particular cases, melt reactions might also be uniform and quantitative if the product crystallizes under the reaction conditions. The essential crystallographic condition for molecular solid-state reactivity is anisotropic molecular migration within the crystals for removal of the enormous local internal pressure as created by the change in molecular shape. Cleavage planes, channels, or voids must enable these migrations. If anisotropic migration is not possible, the phase rebuilding mechanism cannot start and there will be no chemical reaction.33 The creation of internal pressure by the chemical change has been overlooked in previous hypotheses claiming that molecules would not move within crystals, whereas very distinct long-range anisotropic molecular migrations are easily and consistently shown by nanoscopic atomic force microscopy and correlated to the crystal packing.1,33 Any obstacles by difficulties with transformation to the product phase and/or crystal disintegration by separation of the phases (e.g. surface passivation) can be detected with AFM and then engineered by the choice of suitable experimental conditions. Therefore, numerous solid-state reactions that previously failed or did not provide rapid and complete performance, should be taken up again in the light of the exhaustive knowledge from the “phase rebuilding mechanism”. Solid–solid reactions of supramolecular crystals require continuous achievement of new crystal contacts by mechanical agitation, but no “mechanochemistry” (no unselective cleavage of sigma bonds). Only rarely will the molecular mechanism be preceded by mechanochemistry that is mechanical cleavage of covalent bonds. Examples are the milling of the polymer paraformaldehyde or of elementary sulfur in solid-state reactions. Efficient ball milling of polymers produces detectable radicals. Explosives or radical chain starters should not be milled for security reasons. Very different results ensue by milling of 3-D covalent structures such as SiO2, TiO2, Al2O3, or Si, etc. This creates tribochemistry (mechanochemical) involving short-lived triboplasma at the mechanically broken surfaces. In the presence of organic materials the latter are completely mineralized (to give graphite, water, HCl, etc). Tribochemical plasmas can be used to remove all organics from methane to environmental poisons such as benzene and tetrachlorodibenzodioxine (TCCD), etc, in large mills.16,37 These reactions are principally different from the waste-free chemical production by stoichiometric milling of supramolecular crystals as described here. The clear-cut mechanistic differentiation is important for the further development of the field that shall be very important for the sake of a better environment.Solid-state chemical production appears very worthy of being emphasized. The single products obtained do not require purifying workup when starting with pure crystalline compounds and their prior dissolution in solvents for waste-producing liquid-state reactions should be avoided whenever possible.
References
- G. Kaupp, Solid-state molecular syntheses: complete reactions without auxiliaries based on the new solid-state mechanism, CrystEngComm, 2003, 5, 117–133 RSC.
- G. Kaupp, Organic Solid-State Reactions with 100% Yield, Top. Curr. Chem., 2005, 254, 95–183 CAS.
- G. Kaupp and M. R. Naimi-Jamal, Mechanically induced molecular migrations in molecular crystals, CrystEngComm, 2005, 7, 402–410 RSC.
- H. Zoz, H. Ren and N. Späth, Improved Ag-SnO2 electrical contact material produced by mechanical alloying, Metall, 1999, 53, 423–428 Search PubMed.
- R. M. Davis, B. McDermott and C. C. Koch, Mechanical Alloying of Brittle Materials, Metall. Trans. A, 1988, 19, 2867 Search PubMed.
- H. Zoz, H. U. Benz, G. Schäfer, M. Dannehl, J. Krüll, F. Kaup, H. Ren and R. Reichardt, High kinetic processing of enamel, part 1-a, Interceramics, 2001, 50, 388–395 Search PubMed.
- H. Zoz, D. Ernst, R. Reichardt, H. Ren, T. Mizutani, N. Nishida and H. Okouchi, Simoloyer CM100s: semi-continuous mechanical alloying on a production scale using cycle operation – part II, Mater. Manufact. Processes, 1999, 14, 861–874 Search PubMed.
- H. Zoz, D. Ernst, H. Weiss, M. Magini and C. Powell, Mechanical alloying of Ti-24Al-11Nb (at%) using the Simoloyer, Metall, 1996, 50, 575–579 Search PubMed.
- H. Zoz, H. U. Benz, K. Hüttebräucker, L. Furken, H. Ren and R. Reichardt, Stellite bearings for liquid Zn-/Al-system with advanced chemical and physical properties by MA, Metall, 2000, 54, 650–659 Search PubMed.
- H. Zoz, D. Ernst, T. Mizutani and H. Okouchi, Semi-continuously mechanical alloying in a production scale using cycle operation. Part 1, Metall, 1997, 51, 568–572 Search PubMed.
- H. Zoz, D. Ernst, R. Reichardt, T. Mizutani, M. Nishida and H. Okouchi, Semi-continuously mechanical alloying in a production scale using cycle operation. Part 2, Metall, 1998, 52, 521–527 Search PubMed.
- H. Zoz, G. Kaupp, H. Ren, K. Goepel and M. R. Naimi-Jamal, Recycling of EAF dust by semi-continuous high kinetic processing, Metall, 2005, 59, 293–296 Search PubMed.
- G. Kaupp and A. Herrmann, Waste-free quantitative gas/solid diazotization using nitrogen dioxide and triazene synthesis, both avoiding liquid phases, J. Prakt. Chem., 1997, 339, 256–260 CrossRef CAS.
- G. Kaupp, U. Pogodda and J. Schmeyers, Gas/Solid Reactions with Acetone, Chem. Ber., 1994, 127, 2249–2261 CrossRef CAS.
- G. Kaupp, Quantitative large-scale synthesis without auxiliaries for sustainable production, Internat. Chem. Congress of Pacific Basin Societies 2005 – Organic Solid State Chemistry: Structure, Synthesis and Reactivity (ORGN #323), lecture no. 688.
- G. Kaupp, M. R. Naimi-Jamal, H. Ren and H. Zoz, Dry and reactive: Reactive dry-milling for environmental protection, PROCESS Worldwide, 2003, 4, 24–27 Search PubMed , for a long version see http://www.process-worldwide.com/fachartikel/pw_fachartikel_541233.html.
- G. Kaupp, D. Matthies and C. de Vrese, Kohlensäurehalbestersalze durch ressourcenschonende Gas/Festkörper-Addition, Chem.-Ztg., 1989, 113, 219–220 Search PubMed.
- G. Kaupp, Kohlensäurehalbester-Salze, Merck Spectrum, 1991, 3, 42–45 Search PubMed.
- S. Nakatsuji, A. Takai, M. Mizumoto, H. Anzai, K. Nishikawa, Y. Morimoto, N. Yasuoka, J. Boy and G. Kaupp, Preparation and Magnetic Properties of Novel CT Complexes Derived from Organic Stable Radicals, Mol. Cryst. Liq. Cryst., 1999, 334, 177–184 CrossRef CAS.
- G. Kaupp, J. Schmeyers, M. R. Naimi-Jamal, H. Zoz and H. Ren, Reactive milling with the Simoloyer®: environmentally benign quantitative reactions without solvents and wastes, Chem. Eng. Sci., 2002, 57, 763–765 CrossRef CAS.
- G. Kaupp, Verfahren zur Umsetzung von Ketonen mit Stickstoffbasen oder deren Salzen, Ger. Offen, ( 1993), DE 4129757 A1 (to Carl v. Ossietzky Universität Oldenburg, Germany) Search PubMed.
- R. S. Varma, R. Dahiya and S. Kumar, Clay catalyzed synthesis of imines and enamines under solvent-free conditions using microwave irradiation, Tetrahedron Lett., 1997, 38, 2039–2040 CrossRef CAS.
- P. T. Anastas, L. G. Heine and T. C. Williamson, Green Chemical Syntheses and Processes, ACS Symposium Series, Oxford University Press, 2000, vol. 767, p. 298 Search PubMed.
- G. Kaupp, Green Chemical Syntheses and Processes, Angew. Chem., Int. Ed., 2001, 40, 4508–4511 CrossRef.
- G. Kaupp, J. Schmeyers and J. Boy, Quantitative solid-state reactions of amines with carbonyl compounds and isothiocyanates, Tetrahedron, 2000, 56, 6899–6911 CrossRef CAS.
- G. Kaupp, J. Schmeyers and J. Boy, Waste-free solid-state syntheses with quantitative yield, Chemosphere, 2001, 43, 55–61 CrossRef CAS.
- V. P. Chuev, L. A. Lyagina, E. Y. Ivanov and V. V. Boldyrev, Mechanochemical synthesis of phthalazol, Dokl. Akad. Nauk SSSR, 1989, 307, 1429–1432 CAS.
- G. Kaupp and J. Schmeyers, Gas/solid reactions of aliphatic amines with thiohydantoins: atomic force microscopy and new mechanisms, Angew. Chem., Int. Ed. Engl., 1993, 32, 1587–1589 CrossRef.
- J. Schmeyers and G. Kaupp, Heterocycles by cascade reactions of versatile thioureido-acetamides, Tetrahedron, 2002, 58, 7241–7250 CrossRef CAS.
- G. Kaupp, J. Schmeyers and J. Boy, Iminium salts in solid-state syntheses giving 100% yield, J. Prakt. Chem., 2000, 342, 269–280 CrossRef CAS.
- G. Kaupp, F. A. Amer, M. A. Metwally and E. Abdel-latif, Versatile 2-aminothiazoles, building blocks for highly functionalised heterocycles, J. Heterocycl. Chem., 2003, 40, 963–971 CrossRef CAS.
- G. Kaupp, M. R. Naimi-Jamal and J. Schmeyers, Solvent-free Knoevenagel condensations and Michael additions in the solid state and in the melt with quantitative yield, Tetrahedron, 2003, 59, 3753–3760 CrossRef CAS.
- G. Kaupp, Prediction of reactivity in solid-state chemistry, in Making Crystals by Design, ed. D. Braga and F. Grepioni, Wiley-VCH, Weinheim, 2006, pp. 87–147, ISBN: 3-527-31506-2 Search PubMed.
- G. Kaupp and C. Seep, Diastereospecific gas/solid additions with cholesterol derivatives, Angew. Chem., Int. Ed. Engl., 1988, 27, 1511–1512 CrossRef.
- G. Kaupp and A. Kuse, 100% yield in 300 gas–solid reactions covering 22 reaction types: a benign option for industrial solid-state production, Mol. Cryst. Liq. Cryst., 1998, 313, 361–366 CrossRef CAS.
- G. Kaupp, M. R. Naimi-Jamal and V. Stepanenko, Waste-free solid-state protection of diols, diamines, amino acids and polyols with phenylboronic acid, Chem.–Eur. J., 2003, 9, 4156–4161 CrossRef CAS.
- G. Kaupp and H. Zoz, Process for decontamination and/or detoxification of environmental toxicants, e.g. dioxins, dibenzofurans and their congeners, Ger. Offen, 2004, DE 10261204 A1 (to ZOZ GmbH, Germany) Search PubMed.
| This journal is © The Royal Society of Chemistry 2006 |