Detection of trace materials with Fourier transform infrared spectroscopy using a multi-channel detector
K. L. Andrew
Chan
and
Sergei G.
Kazarian
*
Department of Chemical Engineering, Imperial College London, UK SW7 2AZ. E-mail: s.kazarian@imperial.ac.uk
First published on 25th November 2005
Abstract
FTIR spectroscopy is one of the most powerful methods for material characterization. However, the sensitivity of this analytical tool is often very limited especially for materials with weak infrared absorption or when spectral bands of the targeted trace material overlap with the spectral bands of major components. Fortunately, for heterogeneous samples, there is an opportunity to improve the sensitivity of detection by using an imaging approach. This paper explores the opportunity of enhancing the sensitivity of FTIR spectroscopy to detect trace amounts of materials using the FTIR imaging approach based on a focal plane array (FPA) detector. Model sample tablets of ibuprofen in hydroxypropyl methylcellulose (HPMC) have been used to exemplify the detection limits of FTIR spectroscopy using: (a) a conventional mercury cadmium telluride (MCT) detector and (b) a FPA detector. The sensitivity level was compared and it has been found that for this particular set of samples, the lowest concentration of ibuprofen in HPMC that can be detected using attenuated total reflection (ATR) measuring mode with the single element MCT detector was 0.35 wt% while using the FPA detector, the presence of drug has been detected in a sample that contains as little as 0.075 wt% of drug. The application of using this enhanced sensitivity offered by the multi-channel detector to probe trace amounts of drug particles left on the surface of a finger after handling a small amount of the drug has also been demonstrated. These results have broad implications for forensic, biomedical and pharmaceutical research.
Introduction
Analytical detection of trace amounts of material is important especially for applications in food and pharmaceutical industries, forensic science and crime prevention and security measures. Vibrational spectroscopic methods such as Fourier transform infrared (FTIR) spectroscopy are arguably the most powerful techniques available for chemical identifications due to the inherent chemical specificity of these methods. However, the sensitivity of FTIR spectroscopic measurements is often limited by the signal to noise ratio (SNR) especially when the spectral absorbance of the component of interest is weak. SNR can be used to determine the sensitivity limit of a system. If a component gives a spectral feature of 3 times as intense as the noise (SNR > 3), it should be considered as detected. However, for quantitative purposes, the spectral feature should have a SNR of greater than 10 to offer a reasonably small error in the analysis. For the single element detector without using a microscope, the SNR achieved within time, t, can be calculated by the following equation1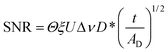 | (1) |
The equation states that SNR is proportional to the throughput (Θ), spectrometer efficiency (ξ), spectral energy density (U), spectral resolution (Δν), detector sensitivity (D*) and square root of t (number of co-additions) and inversely proportional to the square root of the detector area (AD).
Apart from reducing the noise, SNR can also be improved by increasing the signal. The signal level is usually low for the trace material because the Beer–Lambert law states that the infrared absorbance (the signal) is proportional to concentration. Equally, it also states that the absorbance is proportional to the pathlength therefore SNR can be increased by using a longer pathlength. This approach, however, may fail if the infrared absorption bands of the trace material are close to, or overlapping with, the absorption of major components. On the other hand, if the trace materials are heterogeneously distributed in the sample, there are opportunities to increase the sensitivity of detection using microscopy which limits the measured sampling area with an optical aperture. In this case, the sensitivity of the FTIR measurement will not only depend on the SNR but on the spatial resolution as well.
A conventional FTIR spectroscopic measurement using a single element detector collects the averaged signal from the whole sampled area. The weak signal from the trace material is often overwhelmed by the signal from other areas of the sample. In FTIR microscopy, the signal from the sample will be limited to a small area which increases the chance of detecting the trace material by enhancing the spectral signal of the trace material that has a relatively high local concentration. However, the locations of the trace material are usually not known, hence mapping over a large area of the sample is needed in order to detect the trace material. Furthermore, when an aperture is used to increase the spatial resolution of the system, the SNR decreases because the throughput of infrared light, Θ, is reduced. The use of a bright synchrotron source of infrared radiation provide improvement in the throughput but it still requires mapping of the sampling area since the advantages of this source when measuring larger areas are lost. The recently developed focal plane array (FPA) detector can be the method of choice for this application due to its capability to acquire a large number of spectra from a relatively large area of the sample in a short time (within a few seconds). The broad range of applications of FTIR imaging utilising a FPA detector has been demonstrated recently.2–12 The main advantage of FTIR imaging in attenuated total reflection (ATR) mode13 lies in the inherently small pathlength which is suitable for most solid and aqueous samples and the requirement of minimal sample preparation. The introduction of FTIR imaging with a diamond ATR accessory7 extends the application of FTIR imaging to in situ tablet compaction6,14 drug release,9 tablet dissolution studies15,16 and in situ imaging of processes under high pressure gases.10 Recently, we have shown the detection of a trace amount (<0.5%) of component in a tablet using the ATR-FTIR imaging approach.6
The same expression of theoretical SNR (eqn (1)) applies to the FPA detector used in the macro chamber. A reduction in spectrometer efficiency is expected with the FPA detector as there are additional optical components (e.g. mirrors) involved in the imaging measurement. On the other hand, the size of each pixel of the FPA detector is much smaller (40 × 40 µm2) compared to a conventional single element detector (e.g. 1 mm2) used in FTIR spectroscopy and, therefore, the theoretical SNR measured using both detectors should be comparable. However, the electronic design of the FPA detector is far more complex than the single element detector which induces additional noise that would not be found in a single element detector.17 As a result of this, each individual detector pixel in the FPA is expected to have a lower value of detector sensitivity (D*) when compared to the single element mercury cadmium telluride (MCT) detector. Furthermore, the total area of the FPA detector is greater than a single element detector, so the spectral energy density that arrives at each detector pixel is lower. Hence, the SNR level is expected to be much lower for the FPA detector. Nevertheless, the new generation of FPA detectors allows a shorter image acquisition time giving the possibility of spectral quality improvement via the means of image co-addition.
In this paper, the detection limits of FTIR measurements using a single element MCT detector and a FPA detector have been compared. Specifically, the objective of this work was to compare results from both approaches to identify the lowest limit of concentration for the detection of a model substance (drug). The choice of drug for testing sensitivity limits is relatively straightforward because many drugs are encapsulated in excipients in rather small quantities for pharmaceutical applications while detection of trace amounts of illicit drugs also represents a sensitivity challenge. A model system, ibuprofen in HPMC, was prepared via mechanical mixing and weight concentrations ranged from 0.075–1.6% for the purpose of testing the detection limit using the two spectroscopic approaches.
Experimental
Sample preparation
Ibuprofen was supplied by Whitehall International and HPMC was supplied by Colorcon (Orpington, Kent, UK). All samples were used without further purification. Different weight compositions of ibuprofen in HPMC powder were created by mechanically mixing the weighed powder (total mass of ca. 0.5 g) in a beaker with a spatula for a few minutes. Since the mixing is gentle, no change to particles size is expected. The mixtures were stored in a cool dry place until used.FTIR spectroscopy
A single element detector (liquid nitrogen cooled MCT) was used to measure FTIR spectra with an Equinox 55 spectrometer (Bruker Optics) and a modified diamond ATR accessory (“Supercritical fluid analyser”, Specac Ltd.).14 Spectra were collected with 4 cm−1 spectral resolution and 32 scans. A FPA detector (Santa Barbara, USA) is used to measure all FTIR images in conjunction with a macro chamber extension with the same diamond ATR accessory. The arrangement of the optics of this ATR imaging approach is described in more detail in our previous work.7 The FPA detector comprises of 4096 small pixels arranged in a 64 × 64 grid format with each of the pixels measuring an infrared spectrum, thus 4096 spectra are acquired in a single measurement. A plot of the distribution of the absorbance of a specific spectral band corresponding to a particular substance in the imaged area of the sample produces the chemical image for that particular substance. The imaging area measured with this accessory and this particular FPA detector was ca. 530 × 740 µm2. The spectrometer was operated in continuous scan mode, and the spectral resolution and the number of scans were maintained the same as the single element detector measurement which provided a comparable measurement time of ca. 30 s for both methods. Images with a larger field of view were measured using a single reflection ATR accessory with an inverted ZnSe pyramid crystal (Oil analyser, Specac Ltd.).Results and discussions
Ibuprofen/HPMC blend
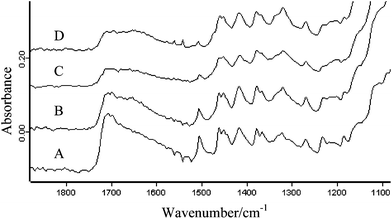
The sample powder mixtures were compacted into tablets of 3 mm diameter with a force of ca. 730 N (ca. 100 MPa) in the diamond ATR accessory and measurements were carried out in situ. The whole diamond ATR accessory was transferred between the sample compartment of the FTIR spectrometer where the conventional FTIR spectrum using the single element detector has been measured and the macro chamber where FTIR imaging spectra were measured using the FPA detector; this was achieved without removing the compacted tablet from the accessory. Fig. 1 shows the fingerprint region of pure ibuprofen and HPMC infrared spectra. The absorption band of HPMC at 1635 cm−1 corresponds to the OH bending mode vibration of water absorbed in HPMC. Ibuprofen has distinct absorption between 1750–1650 cm−1 due to the carbonyl stretching vibration (ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)) which can be used for its characterisation in the blend. The FTIR spectra of HPMC tablets containing trace amounts of ibuprofen (1.6 wt%, 1 wt%, 0.35 wt% and 0.075 wt%) measured using the single element detector MCT are shown in Fig. 2. The peak to peak noise level of the spectra was estimated to be 6 × 10−4 and the peak to peak SNR of the carbonyl band of ibuprofen for the samples containing 1.6 wt%, 1 wt% 0.35 wt% and 0.075 wt% were estimated to be 10, 7, 4 and 0 respectively. Although the SNR for the samples containing 1 wt% and 0.35 wt% were above the detection criteria of 3, the absorption band overlaps with the residual presence of uncompensated water vapour absorption in the background which further degrades the spectral quality. Nevertheless the detection limit for the single element detector with this set up was ca. 0.35 wt% for this system while for quantitative analysis, the detection limit was 1.6 wt%. Below 0.35 wt%, the measurements failed to detect the presence of ibuprofen in HPMC.
O)) which can be used for its characterisation in the blend. The FTIR spectra of HPMC tablets containing trace amounts of ibuprofen (1.6 wt%, 1 wt%, 0.35 wt% and 0.075 wt%) measured using the single element detector MCT are shown in Fig. 2. The peak to peak noise level of the spectra was estimated to be 6 × 10−4 and the peak to peak SNR of the carbonyl band of ibuprofen for the samples containing 1.6 wt%, 1 wt% 0.35 wt% and 0.075 wt% were estimated to be 10, 7, 4 and 0 respectively. Although the SNR for the samples containing 1 wt% and 0.35 wt% were above the detection criteria of 3, the absorption band overlaps with the residual presence of uncompensated water vapour absorption in the background which further degrades the spectral quality. Nevertheless the detection limit for the single element detector with this set up was ca. 0.35 wt% for this system while for quantitative analysis, the detection limit was 1.6 wt%. Below 0.35 wt%, the measurements failed to detect the presence of ibuprofen in HPMC.
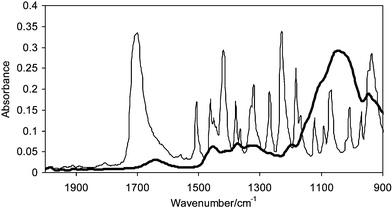 | ||
| Fig. 1 Infrared spectra of ibuprofen (thin line) and HPMC (thick line). | ||
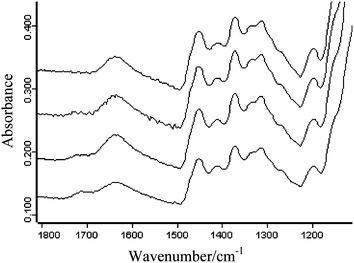 | ||
| Fig. 2 Infrared spectra of sample tablets containing (starting from the top) 0.075 wt%, 0.35 wt%, 1 wt% and 1.6 wt% of ibuprofen in HPMC measured using the single element MCT detector. | ||
FTIR imaging
The same samples were also measured using the FPA detector. FTIR images representing the distribution of ibuprofen in the HPMC matrix were created by plotting the integrated values of the carbonyl band of ibuprofen in the range of 1750–1650 cm−1 over the whole imaged area. These images are shown in Fig. 3 which show that using the FTIR imaging approach, the presence of ibuprofen was detected even when the ibuprofen concentration drops to 0.075%. This was achieved due to the high local concentration of ibuprofen in a certain area within the field of view of the imaging measurement. However, for the sample containing 0.075 wt% of ibuprofen, the drug particles were only detected after the measurement of three tablets with no presence of drug detected in the first two images. The spectra extracted from the ibuprofen-rich domains are shown in Fig. 4. The noise level for the FPA measurement was estimated to be 4 × 10−3, which gives a SNR for the 1.6%, 1%, 0.35% and 0.075% samples of 37, 23, 12 and 10, respectively. The enhanced SNR from the FPA measurements was due to the increase in signal (ibuprofen absorption band) received from the localised area of high ibuprofen concentration although the absolute peak to peak noise level was actually greater than that measured with a single element MCT detector.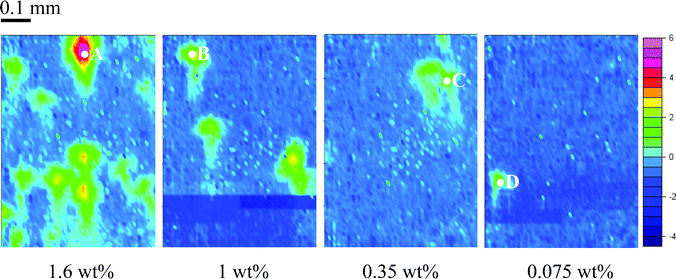 | ||
| Fig. 3 FTIR images showing the distribution of ibuprofen in the compacted tablet with different drug concentrations. The scale bar on the right shows the different colour which represent different integrated absorbance values. The image size is approximately 0.5 mm × 0.7 mm. | ||
 | ||
| Fig. 4 Infrared spectra extracted from the areas indicated in the images in Fig. 3. | ||
Imaging not only offers better sensitivity for heterogeneous substances, it also reveals the distribution of the trace material in the bulk matrix. This is particularly important in resolving issues that arise in the production of pharmaceutical products.7,18 From the images, one can estimate that the domain size of ibuprofen is approximately 100 µm. However, the spectra extracted from the domains with localised ibuprofen concentration were not of pure ibuprofen which suggests that the actual particle size of the drug or matrix was much smaller (indicated by the presence of absorbance of surrounding material). The macro ATR imaging has a reasonable but not particularly high spatial resolution which is on the order of 15–20 µm.7 FTIR imaging with higher spatial resolution (e.g. micro ATR, as demonstrated in ref. 6) may be able to reveal the true drug particle size but this is beyond the scope of this paper. The number of drug domains shown in the images decreases as the concentration of drug decreases. For the lowest concentration of drug, as mentioned before, some of the imaging measurements did not detect any drug domains. This depends on how the drug was distributed in the tablet. Hence, more than one image may be required to be measured when the concentration of the drug is low. If the domain size of the drug particle is larger than the spatial resolution limit of the system, there will be no limit of detection because the drug domains will be detected eventually given that an infinite number of measurements are allowed. However, in reality, the number of measurements is limited by the time available and the cost of the measurement (labour and equipment time). On the other hand, if the spatial domain of the drug is smaller than the spatial resolution, then the sensitivity will still be limited by SNR.
A comparison of single element detector to FPA
Consider the measurement of a trace material in a matrix as shown in Fig. 5, where the trace material would have a 0.5 absorption unit (au) if measured in pure form. If the domain size of trace material is 1/4000th (0.025 vol%) of the size of the measured area, the absorbance of the trace material measured using the single element detector will be averaged to ca. 0.5/4000 and with the criteria of SNR > 3, the maximum noise level allowed for the detector of that material will be ca. 4 × 10−5 au. With the set-up presented in this study, the noise level was ca. 6 × 10−4 with 32 scans. The noise level for a measurement with just 1 scan should be approximately 0.0034 au according to eqn (1). To reduce the noise level to 4 × 10−5 would require 85 fold improvement which might be achieved with ca. 7200 scans if other sources of noise that cannot be removed by co-addition are ignored. Such a noise level is difficult to achieve with the exception of using a synchrotron source of infrared radiation, which is however not easily accessible. On the other hand, when the same sample is measured using the FPA detector, the trace material will be detected by only 1–2 pixels where the absorbance of the trace material will be around 0.5 au. Hence the maximum noise level allowed for the FPA system to detect this trace material will be ca. 0.17 au. This can be easily achieved as the noise level with the FPA detector observed in this study was ca. 4 × 10−3 au.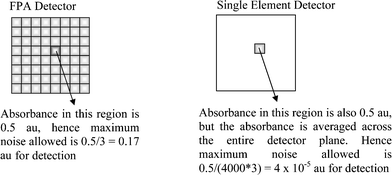 | ||
| Fig. 5 Schematic comparing an absorbance of similar magnitude but measured via the two different approaches. | ||
Forensic science application
Another interesting application of utilising the enhanced sensitivity to detect trace materials by means of FTIR imaging with the FPA detector is in forensic science. A model study to demonstrate this application was performed by an attempt to detect a trace amount of drug particles left on a finger after drug handling. Caffeine has been used as a model drug. A greater field of view is beneficial for this analysis since the surface area of the finger is relatively large, hence increasing the chance of detecting the scattered caffeine particles. The larger sampling area, however, will result in a decrease in spatial resolution. It is often beneficial to measure the sample with different field of views and spatial resolutions by combining different imaging approaches in order to increase the chance of successfully detecting the trace material. As a first attempt to detect the caffeine particles, the ZnSe ATR accessory is employed for this analysis. The surface of a finger was brought in contact with a small amount of caffeine powder (ca. 0.5 mg) followed by wiping the finger with ordinary tissue. The FTIR image was then measured on the surface of the finger to detect any residual caffeine powder. Fig. 6a shows the resultant image of the finger based on the distribution of protein (Amide II at 1535 cm−1). Ridges of the finger print have been revealed clearly. The distribution of caffeine on the finger has been analysed by plotting the absorbance of the caffeine carbonyl band at 1692 cm−1. The resultant image of the caffeine distribution is shown in Fig. 6b. Three small clusters of caffeine with domain size of ca. 300 µm in diameter were detected. Representative spectra from the protein area and the caffeine cluster area were extracted and compared in Fig. 6. The spectrum extracted from the caffeine domain showed a small caffeine band at 1692 cm−1 which represents the presence of caffeine in that area. However, the spectrum is not of pure caffeine, which suggests that the actual caffeine particle size is smaller than the spatial resolution limit of this imaging approach. FTIR imaging with a higher spatial resolution is also available but as mentioned previously, the area measured will be significantly smaller. In that case, several image acquisitions at different areas of the sample may be needed before the drug particles can be detected. However, this problem becomes less significant with the increasing size and speed of FPA detectors. Nevertheless, in this imaging experiment, the presence of a trace amount of caffeine particles on the finger was detected using the FPA imaging system. It is important that detection of these trace amounts of model drug has been achieved with simultaneous measurement (which took only 30 s) of the chemical image of the surface of the finger which makes it powerful case as a forensic evidence. It should also be noted that detection of such small amounts of the model drug deposited on the surface was impossible with conventional FTIR spectroscopy using a single element detector due to limitation of the sensitivity in ATR-FTIR approach.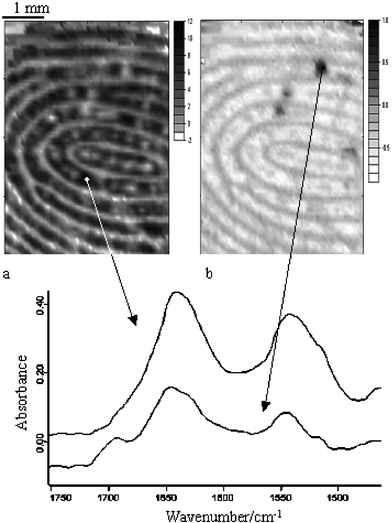 | ||
| Fig. 6 FTIR image of the finger surface. The image on the left (a) represents the protein distribution while the image on the right (b) represents the caffeine distribution. The spectra below the images are extracted from the specific locations of the image which are indicated by the arrows. | ||
Conclusions
The sensitivities of ATR-FTIR measurements with both the single element detector and FPA detector have been studied and compared using model samples. The detection limit was found to be significantly better with the FPA detector for heterogeneous samples due to the increase in signal received from the localised area of high concentration of the trace material. This was achieved because FTIR imaging measures more than 4000 spectra simultaneously within 30 s. However, if the trace material is distributed in the sample homogeneously, an FTIR spectrometer with the single element detector will have a higher sensitivity due to the higher SNR. The application of conventional mapping approach using FTIR microscopy is often impractical due to long acquisition times and sample preparation or impossible with the samples of complex geometry, such as the finger. The demonstrated ATR-FTIR imaging approach provides a non-destructive method to detect trace materials in a wide range of samples that are not amenable for conventional FTIR microscopy.For heterogeneous samples, the advantages of FTIR imaging over the single element detector are not limited to the enhancement of sensitivity. The imaging approach also provides spatial information of the trace material in the matrix such as the distribution and domain size. This is particularly important for the pharmaceutical industry as components distribution in tablets and formulations are crucial to the dissolution properties. The application of FTIR imaging to detect trace amounts of drug particles left on a finger surface after drug handling has been demonstrated. It has been shown that it is possible to detect a small amount of caffeine particles (<300 µm in diameter) that were left on the finger after handling a small amount of the drug. The enhanced sensitivity of FTIR imaging combined with the ATR approach makes this method very powerful for detection of small amounts of particular materials which are heterogeneously distributed in a matrix. For example, the implication of this may impact FTIR imaging studies of the distribution of lipoproteins in medical tissues, such as blood vessels. Indeed, although the total lipoprotein concentrations may be low, its heterogeneous distribution could result in certain small areas of localised high concentration which would be detected by the FTIR imaging approach. This feature of FTIR imaging would make many samples (pharmaceuticals, forensic, medical, etc.) amenable for analytical investigations.
Acknowledgements
We thank EPSRC and Pfizer for support. We thank Mr K. W. T. Kong and Ms D. Vega Escobar for their help.References
- P. R. Griffiths and J. A. de Haseth, Fourier Transform Infrared Spectrometry, Wiley, New York, 1986 Search PubMed.
- J. L. Koenig, Microscopic Imaging of Polymers, ACS, Washington DC, 1998 Search PubMed.
- C. M. Snively and J. L. Koenig, Macromolecules, 1998, 31, 3753–3755 CrossRef CAS.
- A. J. Sommer, L. G. Tisinger, C. Marcott and G. M. Story, Appl. Spectrosc., 2001, 55, 252–256 CrossRef CAS.
- K. Artyushkova, B. Wall, J. L. Koenig and J. E. Fulghum, J. Vac. Sci. Technol., A, 2001, 19, 2791–2799 CrossRef CAS.
- K. L. A. Chan, S. V. Hammond and S. G. Kazarian, Anal. Chem., 2003, 75, 2140–2147 CrossRef CAS.
- K. L. A. Chan and S. G. Kazarian, Appl. Spectrosc., 2003, 57, 381–389 CrossRef CAS.
- K. L. A. Chan and S. G. Kazarian, J. Comb. Chem., 2005, 7, 185–189 CrossRef CAS.
- S. G. Kazarian and K. L. A. Chan, Macromolecules, 2003, 36, 9866–9872 CrossRef CAS.
- S. G. Kazarian and K. L. A. Chan, Macromolecules, 2004, 37, 579–584 CrossRef CAS.
- B. A. Miller-Chou and J. L. Koenig, Macromolecules, 2002, 35, 440–444 CrossRef CAS.
- K. L. A. Chan, S. G. Kazarian, A. Mavraki and D. R. Williams, Appl. Spectrosc., 2005, 59, 149–155 CrossRef CAS.
- E. M. Burka and R. Curbelo, US Pat. 6141100, 2000.
- K. L. A. Chan, N. Elkhider and S. G. Kazarian, Chem. Eng. Res. Des., 2005, 83, 1303–1310.
- J. Van der Weerd and S. G. Kazarian, J. Controlled Release, 2004, 98, 295–305 CrossRef.
- J. Van der Weerd, K. L. A. Chan and S. G. Kazarian, Vib. Spectrosc., 2004, 35, 9–13 CrossRef.
- C. M. Snively and J. L. Koenig, Appl. Spectrosc., 1999, 53, 170–177 CrossRef CAS.
- Y. Roggo, A. Edmond, P. Chalus and M. Ulmschneider, Anal. Chim. Acta, 2005, 535, 79–87 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |