A green and facile route to γ- and δ-lactones via efficient Pinner-cyclization of hydroxynitriles in water†
Karolina
Aplander
,
Olle
Hidestål
,
Kambiz
Katebzadeh
and
Ulf M.
Lindström
*
Organic Chemistry, Lund University, P. O. Box 124, SE-221 00 Lund, Sweden. E-mail: ulf.lindstrom@organic.lu.se; Fax: +46 46 2228209; Tel: +46 46 2228211
First published on 23rd November 2005
Abstract
In the presence of a recyclable cationic exchange resin hydroxynitriles smoothly undergo a Pinner cyclization/hydrolysis two-step reaction in pure water to give lactones in good to excellent yields.
Lactones are frequently occurring structural subunits in many biologically active natural products, for example natural flavors and pheromones. Functionalized chiral lactones have found extensive use as building blocks for the stereoselective preparation of alkaloids, antibiotics, pheromones and flavor components.1 It is therefore of importance to develop simple and reliable methods of preparing lactones, and a variety of methods have indeed been introduced, the most common being cyclization of hydroxyacids or haloacids.2 On the other hand, hydroxynitriles should be highly useful as precursors of lactones because of the ease with which various nitriles can be prepared, for example by cyanide ion nucleophilic substitution at electrophilic carbons. Nevertheless, this approach towards lactones has been severely hampered by the harsh conditions usually required. A recent attempt to convert hydroxynitriles into lactones via mild, enzymatic hydrolysis of the cyano group was of limited value for synthetic purposes.3 Thus, a useful method for the direct conversion of hydroxynitriles into lactones is a desirable yet unfulfilled goal. Also, because of environmental concerns and increased restrictions on the use of hazardous organic solvents it has recently become of significant interest to develop reactions in water, which is a cheap, safe and non-toxic solvent.4 If, in addition, aqueous reactions can be efficiently mediated by heterogeneous catalysts that can be recycled and reused many times, the result will be nearly ideal processes in terms of both greeness and simplicity. Herein we present the direct, high-yielding conversion of hydroxynitriles into lactones using water as solvent and a cationic exchange resin as a recyclable, heterogeneous catalyst.
In an ongoing synthetic project we needed to convert a γ-hydroxynitrile into the corresponding lactone. The few reported experiments that exist on this transformation are two-step procedures.5 First, cyclization of the starting hydroxynitrile is achieved by an intramolecular Pinner reaction to give a cyclic imidate (Scheme 1). This step has been shown to require strong acid catalysis in organic media (e.g. HCl/EtOH or HCl/ether),5d,e except in highly favorable cases where aqueous basic,5a or even neutral5b conditions have been used. The second step, hydrolysis of the cyclic imidate into the lactone, typically involves highly acidic aqueous conditions at elevated temperatures.5a,b,d,e
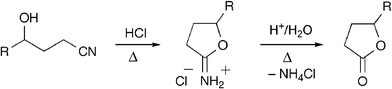 | ||
| Scheme 1 The two-step Pinner-cyclization/hydrolysis reaction sequence has been of limited use in the preparation of lactones due to the harsh conditions usually required. | ||
Not surprisingly, the harsh conditions caused severe problems with side reactions in our synthetic study and it was necessary for us to develop an alternative method. Recently, Mizuno and co-workers reported an efficient procedure for the hydration of nitriles to amides in water using an alumina-supported ruthenium catalyst (Ru(OH)x/Al2O3).6 The reaction was proposed to proceed via coordination of the nitrile to the ruthenium metal. The activated nitrile then undergoes facilitated hydrolysis. We reasoned that if a hydroxynitrile was used, an intramolecular attack of the hydroxyl group could take place to form a cyclic imidate rather than an open chain amide. In a first attempt we heated hydroxynitrile 1a at 125 °C in water in the presence of Ru(OH)x/Al2O3 (5% Ru). This led to complete conversion of starting material, but only after 27 h, and three products were detected. These were identified as the expected cyclic imidate 2a and, gratifyingly, lactone 3a in a 3 ∶ 2 ratio, along with minor amounts of the amide corresponding to the hydrolysis product of nitrile 1a (Table 1, entry 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalysta | X− | Time/h | T/°C | 3a ∶ 2a |
| a 5% Ru was used in entries 1–4. b Also contained minor amounts of amide corresponding to the hydrolysis product of 1a. | |||||
| 1 | Ru(OH)x | OH− | 27 | 125 | 2 ∶ 3b |
| 2 | RuCl3 | OH− | 42 | 135 | 9 ∶ 1 |
| 3 | RuCl3/acetic acid (100%) | AcO− | 42 | 130 | 7 ∶ 3 |
| 4 | RuCl3/Dowex 50 × 8 (H+) | RSO3− | 6 | 135 | 1 ∶ 0 |
| 5 | Dowex 50 × 8 (H+) | RSO3− | 1 | 135 | 1 ∶ 0 |
| 6 | — | OH− | 6 | 135 | 1 ∶ 3 |

The formation of significant amounts of lactone was encouraging at this stage. The presence of amide in the product mixture was not surprising in view of the work by Mizuno and co-workers described above. However, according to their mechanistic proposal, the hydroxide that is added to the cyano group does not originate from the surrounding water but from Ru(OH)xvia an intramolecular transfer. We therefore decided to test the use of RuCl3 a reagent which, although inferior to Ru(OH)x/Al2O3, also converted nitriles to amides but presumably via a different mechanism where the hydroxide must be delivered from a source external to the RuCl3–CN complex. Indeed, heating 1a with RuCl3 (5%) in water in a sealed tube at 135 °C for 42 h yielded no detectable amide, but cleanly converted starting material into lactone 3a and cyclic imidate 2a in a 9 ∶ 1 ratio (entry 2). No other products were observed.
With hopes of reducing reaction time, we decided to add a Brønsted acid as co-catalyst. An initial attempt using acetic acid as additive had no significant effect on the rate of the reaction (entry 3). On the other hand, a remarkable improvement was found when we ran the reaction in the presence of a solid-supported acid catalyst. Heating 1a with RuCl3 (5%) in water with acidic Dowex 50 W × 8–200 (1.7 meq mL−1) cation exchange resin at 135 °C afforded lactone 3a exclusively in only 6 h (entry 4).7 At this point we became interested in deducing the roles played by the metal and the acid catalyst in mediating lactonization, and to initiate such a study we ran the reaction without RuCl3. Surprisingly, the reaction proceeded efficiently also in the absence of metal! In addition, we were able to reduce the reaction time drastically to just one hour (entry 5). No other products were detected and the lactone was obtained in spectroscopic purity (1H-, 13C-NMR) after filtration and removal of the solvent under reduced pressure. When we ran the reaction in neat water, without catalyst, we found to our further surprise that significant amounts of the lactone was formed after 6 h reaction at 135 °C. No starting material was observed and the imidate was the major product (3a/2a 1 ∶ 3, entry 6). From this result we draw the conclusion that the resin plays an important part in accelerating the hydrolysis of the intermediate imidate. A plausible mechanistic rationale is that the charged cyclic imidate becomes localized at the ionic surface of the catalyst, where hydrolysis to lactone should be facilitated because of the highly acidic environment.
The high purity of the crude product can in part be explained by efficient removal of the stoichiometric ammonium ion byproduct from the reaction mixture through salt formation with the sulfonic acid groups of the catalyst resin, which also accounts for the need for more than a stoichiometric amount of catalyst. Unfortunately, attempts at running the reaction at lower temperature were not successful. At temperatures below 100 °C the reaction became extremely sluggish. On the other hand, higher temperatures than 140 °C led to thermal decomposition of the resin.
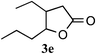
After having established that the use of a cationic exchange resin in water efficiently promotes the projected Pinner cyclization/hydrolysis reaction sequence, we proceeded to investigate the scope of this method by applying it to other hydroxynitriles. As can be seen in Table 2, various γ-hydroxynitriles (1a–1f) cyclize to the corresponding γ-lactones (3a–3e) in good to excellent yields (65–99%) under the described conditions (entries 1–5). Gratifyingly, we were also able to apply this method to the preparation of a δ-lactone. Cyclization of δ-hydroxynitrile 1f under the described conditions gave dihydrocoumarin, 3f, thus suggesting an efficient entry to the attractive coumarin derivatives. A considerably longer reaction time (47 h) was required, however, in order to get a good yield (79%, entry 6). In general, the procedure for isolating the products from the aqueous reaction mixture was limited to removal of the cation exchange resin by filtration and evaporation of the solvent.
| Entry | Hydroxynitrile | Lactone | Time/h | Yield (%)b |
|---|---|---|---|---|
| a For a typical experimental procedure, see ref. 8. b Yields refer to crude products of >95% purity by NMR, except for 3a which was purified by flash chromatography. c Work-up by extraction with diethyl ether. Quantitative recovery of unreacted imidate from the water phase. d Performed with a mixture of diastereomers. Diastereomeric ratio retained in the product. | ||||
| 1 |

|

|
1 | 96 |
| 2 |

|

|
6 | 95c |
| 3 |

|

|
6 | >99 |
| 4 |

|

|
30 | 83c |
| 5d |

|
|
1 | 65 |
| 6 |

|

|
47 | 79 |
Alternatively, in the cyclizations of 1b and 1d, where volatility was a potential problem, lactones 3b and 3d were extracted from the filtrate with diethyl ether (entries 2 and 4). In these cases, the lactones and the water-soluble unreacted imidate intermediates were efficiently separated into the organic and the aqueous phases, respectively, and the material was quantitatively recovered by concentration of the phases. Either work-up procedure led to >95% pure lactones (1H-, 13C-NMR) thus obviating the need for costly and time-consuming chromatography for most synthetic purposes.
Finally, in order to verify that the solid catalyst could be recycled, we recovered the resin from the cyclization of 1c, reactivated it by treatment with a small amount of 1 M HCl and used it in subsequent cyclizations. The reaction was performed three times using the same resin and only a small decrease in the isolated yield of 3c was observed (Scheme 2).
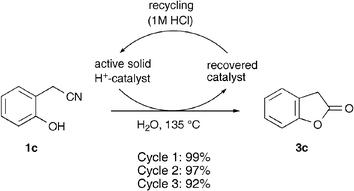 | ||
| Scheme 2 Recovery of the cation exchange resin for recycling and use in subsequent cyclization/hydrolysis reactions. | ||
In conclusion we have developed a useful method of preparing synthetically attractive γ- and δ-lactones via the cyclization of γ- and δ-hydroxynitriles. In comparison with reported protocols for preparing lactones from hydroxynitriles, the method presented herein avoids the use of concentrated mineral acids in organic and aqueous–organic media and instead employs a heterogeneous catalyst that is easily removed from the product mixture, and which can be recycled and reused. No metals or other hazardous chemicals are employed and the reactions are run in pure water, which is the cheapest and most harmless solvent available. Finally, lactones of >95% purity were simply and conveniently obtained by filtration followed by concentration of the filtrate. This is in part due to relatively short reaction times, but also because ionic byproducts can associate strongly with the catalyst itself and efficiently be removed from the reaction mixture, thus greatly facilitating purification procedures. These synthetic as well as green advantages should make cyclization of hydroxynitriles a more attractive route to various lactones.
Acknowledgements
We thank the Swedish Research Council and the Crafoord Foundation for financial support.References
- (a) C. García, T. Martín and V. S. Martín, J. Org. Chem., 2001, 66, 1420 CrossRef CAS; (b) H. Takahata, Y. Uchida and T. Momose, J. Org. Chem., 1994, 59, 7201 CrossRef CAS and references therein.
- For a list of methods, see: M. B. Smith and J. March, Advanced Organic Chemistry, Wiley, New York, 5th edn, 2001, p. 1680 Search PubMed.
- S. K. Taylor, N. H. Chmiel, L. J. Simons and J. R. Vyvyan, J. Org. Chem., 1996, 61, 9084 CrossRef CAS.
- For recent reviews, see: (a) C. J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS; (b) U. M. Lindström, Chem. Rev., 2002, 102, 2751 CrossRef; (c) Organic Synthesis in Water, ed. P. A. Grieco, Blackie Academic & Professional, London, 1998 Search PubMed; (d) C. J. Li and T. H. Chan, Organic Reactions in Aqueous Media, Wiley, New York, 1997. See also thematic issue: “Organic Reactions in Water”, Adv. Synth. Catal., 2002, 344(3–4) Search PubMed.
- For representative examples, see: (a) F. Fringuelli, O. Piermatti and F. Pizzo, J. Chem. Educ., 2004, 81, 874 CrossRef CAS; (b) S. Darvesh, A. S. Grant, D. I. MaGee and Z. Valenta, Can. J. Chem., 1991, 69, 723 CAS; (c) M. J. Fisher, W. J. Hehre, S. D. Kahn and L. E. Overman, J. Am. Chem. Soc., 1988, 110, 4625 CrossRef CAS; (d) R. Kwok and M. E. Wolff, J. Org. Chem., 1963, 28, 423 CrossRef CAS; (e) D. H. R. Barton, J. M. Beaton, L. E. Geller and M. M. Pechet, J. Am. Chem. Soc., 1960, 82, 2640 CrossRef CAS.
- K. Yamaguchi, M. Matsushita and N. Mizuno, Angew. Chem., Int. Ed., 2004, 43, 1576 CrossRef CAS.
- The amount of dry resin used was 10 times the weight of the starting hydroxynitrile, which based on the exchange capacity of Dowex 50 W × 8–200 (2.1 meq g−1) corresponds to approximately 2 equivalents of acid.
- Representative procedure: 2-Coumaranone (3c). To a mixture of 2-hydroxyphenylacetonitrile, 1c (18.8 mg, 0.14 mmol), in H2O (1 mL) was added cationic exchange resin (190 mg, Dowex 50 W × 8–200). The reaction mixture was stirred vigorously in a sealed tube at 135 °C for 6 h. After cooling to room temperature, the catalyst was filtered off and washed with ethanol. The filtrate was concentrated with gentle heating under reduced pressure to give 2-coumaranone, 3c, in quantitative yield. Spectroscopic data were in accordance with commercially available material. Reactions with more volatile molecules can alternatively be worked up by a double extraction of the filtrate with diethyl ether, followed by drying of the combined organic phases over MgSO4 and concentration at reduced pressure.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details and procedures for the preparation of 1e and 3e. 13C-NMR spectra for all lactones. See DOI: 10.1039/b513656c |
| This journal is © The Royal Society of Chemistry 2006 |