Selective catalytic reduction of NO over supported silver catalysts—practical and mechanistic aspects
Ken-ichi Shimizu and Atsushi Satsuma*
Department of Applied Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8603, Japan. E-mail: satsuma@apchem.nagoya-u.ac.jp; Fax: +81-52-789-3193; Tel: +81-52-789-4608
First published on 23rd May 2006
Abstract
Selective catalytic reduction of NO by hydrocarbons (HC-SCR) is one of the promising technologies for removal of NO in exhausts containing excess oxygen, such as diesel and lean burn gasoline engines. Supported Ag catalysts, especially Ag/Al2O3, are thought to be the promising candidates for use in diesel exhausts, as confirmed by several reports on engine bench tests. The HC-SCR performance of supported Ag catalysts is very sensitive to the reaction conditions, especially the type of hydrocarbons and the addition of H2. The control of reaction conditions would be key for practical use. The current research of supported Ag catalysts is reviewed from the viewpoints of practical use and the reaction mechanism, i.e., the reaction scheme, the role of surface adsorbed species, and the structure of active Ag species.
1. Introduction
1.1 Automobiles as sources of NOx emissions
Nitrogen oxides (NOx: NO and NO2) are major air pollutants that cause photochemical smog formation and acid rain. The emission of various nitrogen oxides into our atmosphere occurs on a massive scale. Worldwide, over 30 million tons of NOx are vented to the earth’s atmosphere each year.1 There has been a worldwide effort to discover improved solutions for the removal of NOx emissions from stationary and mobile sources. Among sources of NOx from automobiles, the demands for lean burn gasoline and diesel engines in vehicles are expected to increase because there are strong efforts towards reducing CO2 emissions, according to the Kyoto Protocol to the United Nations Framework Convention on Climate Change.2 The improvement of fuel economy and lower emissions of CO2 is important in automobiles from now on. Fig. 1 shows the total CO2 emissions from various types of automobiles relative to a gasoline engine.3 Among various engine technologies, a gasoline hybrid engine is the most effective technology on the market for the reduction of CO2 emissions. Fuel cell engine cars do not exhaust CO2 itself, although significant amounts of CO2 are emitted from the transportation and production of H2 from fossil fuels such as natural gas or petroleum. As a result, the total CO2 emission is almost equivalent to gasoline hybrid engines. Diesel engines also show higher fuel economy than gasoline engines. Further reduction of CO2 emission can be expected by the use of hybrid technology or bio-diesel fuels.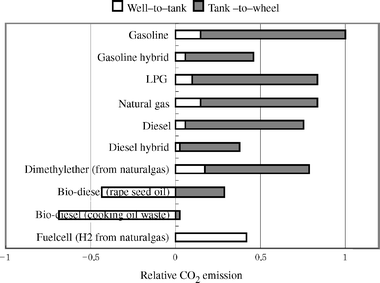 | ||
| Fig. 1 CO2 emission from various vehicles relative to gasoline engine cars. (Reproduced with permission from ref. 3) | ||
Purification of diesel exhaust, i.e., reduction of particulate materials (PM) and NOx, is the essential problem to be solved for energy efficient diesel engines. For gasoline engines, effective de-NOx technology is already established by using a three-way catalyst, which is composed of supported precious metals including Pt, Rh, and Pd.4,5 On the three-way catalysts, NO is reduced to N2 by unburned hydrocarbons (HC), CO, and H2, and HC and CO are oxidized to CO2 simultaneously. The recent development of a CeO2–ZrO2 support significantly advanced the performance of the three-way catalysts.6 Since the air : fuel ratio is higher in lean burn gasoline engines and diesel engines, O2 concentration is around 10% in exhausts. Although the target reaction is the reduction of NO by HC, undesired consumption of HC by combustion with excess O2 occurs. To control these competitive reactions, new catalysis technology based on novel catalysts is desired.
1.2 Development of NOx reduction technology for oxygen-rich exhaust
There are several candidates for de-NOx technology for oxygen-rich exhausts.5,7 For automobile diesel engines, the following three technologies are developing and coming onto the market: diesel particulate and NOx reduction system (DPNR), selective catalytic reduction by urea (Urea-SCR) and selective catalytic reduction by hydrocarbons (HC-SCR). Toyota Motor Co. Ltd has already developed DPNR technology for simultaneous reduction of both NOx and PM from diesel exhausts.8–10 This technology is based on the NOx storage reduction (NSR) catalyst system for lean burn gasoline engines.11,12 The NSR system is performed with an engine that operates alternately under lean or rich conditions and is characterized as a time-resolved HC-SCR over an evolved three-way catalyst. The catalyst contains NOx storage materials, such as BaO, which store NOx as nitrates during lean conditions. The stored NOx is released from the storage materials and reduced over noble metals by hydrocarbons and CO when turning the engine to rich conditions for a short period. In the DPNR system, stored nitrates also oxidize deposited PMs trapped on the wall of ceramic filter. This technology has been applied to commercial passenger cars from the end of 2003.Urea-SCR is thought to be effective for large-scale diesel engine cars such as tracks and buses.13–16 Daimler-Chrysler and Nissan Diesel Motor Co., Ltd. have adopted the Urea-SCR system for commercial tracks.17,18 Daimler-Chrysler has announced that the Mercedes-Benz E 320 with the urea-SCR system named “BLUETEC” will be launched in the fall of 2006 in the US. The use of NH3 is very effective to reduce NOx to N2 by using zeolite-based catalysts or vanadium-based catalysts, but it would not be possible to transport cylinders of NH3 on automobiles. Therefore, aqueous solutions of urea ((NH2)2CO) are used as selective reduction agents. In the presence of H2O, urea is hydroxylated to NH3 and CO2 according to the following equation
| (NH2)2CO + H2O → 2NH3 + CO2. | (1) |
Selective catalytic reduction of NO by hydrocarbons (HC-SCR) in excess oxygen is also thought to be a potential method for removing NOx from lean burn and diesel exhausts. In 1990, Iwamoto et al.19 and Held et al.20 discovered that NOx reduction under excess oxygen could be accomplished using hydrocarbons as the reducing agents over Cu–MFI, i.e., Cu ion-exchanged MFI-type zeolite. The use of unburned hydrocarbons in exhausts as the reductant is the main advantage of the HC-SCR system. Moreover, the HC-SCR system proceeds even in the presence of excess oxygen and the presence of oxygen is indispensable, which is a distinguishing characteristic of this system. Thus, this technology has the possibility of overcoming the disadvantages of the present reduction system, i.e., the three-way catalytic system.
Since the discovery of HC-SCR technology, numerous screening runs have been carried out and various types of HC-SCR catalysts have been reported, such as ion-exchanged zeolites, supported precious metals, and metal oxide-based catalysts, containing various active components such as Cu, Co, Fe, Ag, Pt, Pd, In, Ga, and Sn.5,21–25 Ion-exchanged zeolites, especially Cu–MFI, show very high performance on the HC-SCR. From a practical point of view, however, they have some potential problems originating from the character of zeolites, i.e., instability in hydrothermal conditions that causes deactivation of catalysts. Supported precious metal catalysts, especially supported Pt catalysts, show high stability and high tolerance to sulfur oxides (SOx) and water vapour. They show very high activity at lower temperatures, but the formation of N2O and a narrow temperature range for NOx reduction are problems still to be overcome. Metal oxide-based catalysts shows high stability and moderate tolerance to SOx and water vapour. Amongst a number of catalysts reported, Ag/Al2O3 is thought to be the most promising candidate for practical use. Several reports on engine bench tests demonstrated the potential of Ag/Al2O3 in practical applications,26–31 but low SCR activity at lower temperatures should be improved.
Despite the large number of reports on catalyst developments, the performance of the HC-SCR catalysts has not yet reached a practical level. This implies that there is a limit to the improvement in SCR performance through the design of the catalyst alone. Although most of the tests of HC-SCR catalysts have been carried out at fixed reaction conditions, it is possible to control the reaction conditions of practical automobile catalysts by controlling the engines. The NSR catalyst is an excellent example.5,11,12 The NSR catalyst plays different roles under lean and rich conditions for the effective removal of NOx. A drastic enhancement of the HC-SCR activity can be expected by using the HC-SCR catalysts under unsteady state reaction conditions or periodic operation. For the HC-SCR system, Iwamoto and co-workers proposed the intermediate addition of reductant (IAR) concept,32,33 which is performed using hydrocarbons, such as fuel, injected into the exhaust gas. The injected hydrocarbons act as reducing agents for NO2, which has been formed by the oxidation of NO in the exhaust. Since the concentration of unburned hydrocarbons in the diesel exhaust is relatively lower than in the case of gasoline engines, the IAR method is thought to be a preferable option for the HC-SCR system. The combination of a HC-SCR catalyst and plasma reactor is also effective for simultaneous reduction of NOx and degradation of PMs.34–43 However, the development of active and selective SCR catalysts is still the key technology for the development of these modified HC-SCR systems.
After Miyadera and Yoshida reported the high activity of SCR with ethanol over Ag/Al2O3,44,45 supported Ag catalysts have attracted much attention. The higher hydrocarbons such as octane also act as effective reductants for HC-SCR over Ag/Al2O3.46–49 In fact, good performances of Ag/Al2O3 catalysts have been confirmed by engine bench tests using secondary fuel injection and the use of ethanol as a reductant.26–31 With regard to the catalyst design, the addition of metal additives, such as Pd and Rh, has been reported to be effective for the enhancement of the SCR activity of Ag/Al2O3.50,51 Since real automobile exhausts contain water vapour and SOx, the automobile catalysts should have tolerances to these atmospheres, which act as poisons to the catalysts. Ag/Al2O3 have been reported to have moderate tolerance to water vapour and SO2.28–30,52–58 The low price of Ag is also attractive for practical uses. There have been various scientific investigations on the reaction mechanism and the effective species for HC-SCR. This review summarizes the practical and scientific research on the HC-SCR over supported Ag catalysts.
2. HC-SCR on supported Ag catalysts—strong dependence on reaction atmospheres
2.1 Oxygenated hydrocarbons as reductants
Ag-based SCR catalysts are known to be very sensitive to reaction conditions, especially reducing reagents for NO. In the early research on the HC-SCR catalysts, Ag-based catalysts were thought to be ineffective because Ag–zeolites show only poor activity to the selective reduction of NO by ethylene.22 On the other hand, Miyadera first discovered the high efficiency of Ag/Al2O3 catalysts in NO reduction to N2 by oxygenated hydrocarbons, such as ethanol.44,45,59,60 By using ethanol as a reductant, Ag/Al2O3 catalysts showed very high SCR performance, i.e., nearly 100% of NO conversion at around 623–723 K.44,45 When 2-propanol, acetone, and ethanol were used as reductants for NO, the conversion of NOx achieved above 80% at 523–673 K (SV = 6400 h−1) even in the presence of 10% water vapour. The conversion of NO was significantly affected by the type of oxygenates, i.e., the conversion of NO was in the order of 2-propanol > acetone > ethanol > 1-propanol ≫ methanol at 350 °C, as shown in Fig. 2.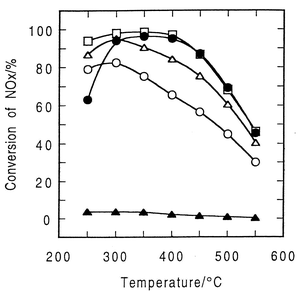 | ||
| Fig. 2 Conversion of NO by oxygen-containing organic compounds over 2 wt% Ag/Al2O3 catalyst. Reaction conditions: 500 ppm NO, 1000 ppm (as CHx) reductant (333 ppm), 10% O2, 10% CO2, N2 balance, 10% H2O, SV = 6400 h−1. Symbols: (○) 1-propanol, (□) 2-propanol, (Δ) ethanol, (●) acetone, and (▲) methane. Reprinted from Appl. Catal., B, 2, T. Miyadera, Alumina-supported silver catalysts for the selective reduction of nitric oxide with propene and oxygen-containing organic compounds, pp. 199–205, © 1993, with permission from Elsevier.45 | ||
The potential of ethanol-SCR over Ag/Al2O3 for practical use has been demonstrated through a diesel engine bench test.30,31,61 He and Yu31 reported that the catalytic converter of Ag/Al2O3 showed an average NOx conversion of greater than 80% in the temperature range of 300–400 °C. In the case of a space-velocity of 30 000 h−1, the light-off temperature was 270 °C and the highest NOx conversion reached 92.3% at 400 °C. Thomas et al.30 tested SCR by various alcohols for the exhaust from 5.9 L diesel engine with 7.0 L Ag/Al2O3 catalyst system with a reductant injector. The light primary alcohols, such as ethanol, 1-propanol and 1-butanol, gave greater than 80% NOx reduction in the temperature range of 300–450 °C and at SV = 50 000 h−1. Recently, Kocat Co. Ltd demonstrated a practical de-NOx system of ethanol-SCR over Ag/Al2O3.61 The critical improvement of the system has been made on the injection of ethanol reductant. The introduction of ethanol reductant into the catalyst bed and exhaust was performed by multiple installations and an injection manner to effectively remove NOx, preventing ethanol from being oxidized midway in the catalyst bed.
2.2 Use of higher hydrocarbons as reductants
Although Ag/Al2O3 shows very high performance on ethanol-SCR, it is difficult to use ethanol on automotive SCR systems because of the inconvenience of attaching an extra tank for ethanol. The secondary fuel injection into exhaust gas, i.e., internal addition of reductant,32,33 is thought to be an effective method for automotive diesel exhausts because an extra tank of reductants is not required and a lower concentration of unburned hydrocarbons than that of NOx in diesel exhausts. When the secondary fuel injection into exhaust was employed, the catalysts are required to effectively use higher hydrocarbons as the reductant. Several researchers have focused on the effect of increasing carbon number 47,62–64 or the use of higher hydrocarbons as the reductant 26,27,46–49,62–67 on various types of SCR catalysts, such as Co–ZSM-5,62 Pt-based catalysts,63–66 Co-based catalysts,26 and Cu/sulfated zirconia.67 The systematic investigation on the effect of hydrocarbon reductants has been carried out by Burch and Millington by using Pt/Al2O3 and Pt/SiO2 catalysts.64 They reported that the SCR efficiency increases in the order i-paraffins < aromatics < n-paraffins < olefins = alcohols and generally activity increases with an increase in carbon number.For Ag/Al2O3 catalysts, the use of higher hydrocarbons is preferable for both the activity and the water tolerance on HC-SCR.5,47,68–70Fig. 3 shows the conversion of NO over 2wt% Ag/Al2O3 prepared by a sol-gel method in the presence of 2% water vapour when various linear alkanes from methane to n-octane are used as a reductant.47 As the carbon number in alkanes increased, the temperature window of NO reduction shifted to lower temperature. Light-off temperatures for hydrocarbon conversion also shifted to lower temperatures with an increase in the carbon number. When n-octane was used as a reductant, sufficient SCR activity was achieved from 550 K. On the other hand, in the absence of water vapour, the n-hexane-SCR showed the higher activity at low temperatures than that of n-octane-SCR, as shown in Fig. 4. This is due to the suppression of the poisoning by deposited carboxylates and carbonate species by water vapour.48 For SCR by n-octane and i-octane in the presence of water vapour, Ag/Al2O3 showed higher NO conversion to N2 and selectivity than Pt/Al2O3 and Cu–ZSM-5. The nature of the Ag/Al2O3 catalyst is thought to be advantageous for HC-SCR of diesel exhaust with secondary diesel fuel injections before the catalyst bed.28,29
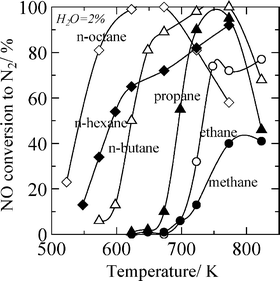 | ||
| Fig. 3 NO conversion to N2 on an Ag/Al2O3 catalyst using various n-alkanes in the presence of water vapor. Conditions: NO = 0 or 1000 ppm, HC = 6000 ppm C, O2 = 10%, H2O = 2%, and W/F = 0.12 g s ml−1 except for methane-SCR (W/F = 0.9 g s ml−1). Reprinted from Appl. Catal., B, 25, K. Shimizu, A. Satsuma and T. Hattori, Catalytic performance of Ag–Al2O3 catalyst for the selective catalytic reduction of NO by higher hydrocarbons, pp. 239–247, © 2000, with permission from Elsevier.47 | ||
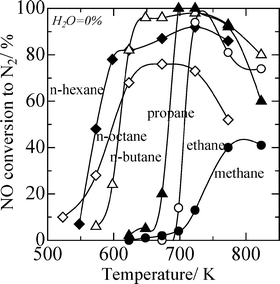 | ||
| Fig. 4 NO conversion to N2 on Ag/Al2O3 catalyst using various n-alkanes in the absence of water vapour. Conditions: NO = 1000 ppm, HC = 6000 ppm C, O2 = 10%, and W/F = 0.12 g s ml−1 except for methane-SCR (W/F = 0.9 g s ml−1). | ||
The advantage of the HC-SCR by higher hydrocarbons can be attributed to the reactivity of hydrocarbons. Shibata et al.70 reported that hydrocarbon structure had an influence on the HC-SCR activity: NO reaction rate increased with an increase in the carbon number in linear alkanes, and the rate in SCR by branched alkanes is lower than that by linear alkanes of the same carbon number. Fig. 5 shows the reaction rates of NO as a function of “mean bond energy” of the hydrocarbons, which is an average of all C–H and C–C bond energies in the hydrocarbons. Good correlations are observed on the HC-SCR by linear alkanes and branched alkanes over Ag/Al2O3, Ag–MFI, Cu/Al2O3 and Cu–MFI. It should be noted that the reaction rate was not dependent on the weakest C–H bond, which often contributes to the rate limiting step and controls the reaction rate in selective oxidation.71 The strong dependence of the HC-SCR activity on the “mean bond energy” means that the HC-SCR reaction takes place with multiple reaction steps concerting rate limiting. The slope of the correlation was greater in Ag/Al2O3 than in the other catalysts, indicating unique behaviour for Ag/Al2O3 which may be correlated to the changeable state of Ag, depending on reaction conditions. The mechanistic cause of these correlations can be rationalized by the reaction rate of the formation of surface oxygenated hydrocarbons, which has been revealed by in situ IR experiments, as reviewed in section 3.2.
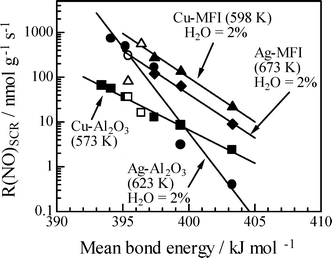 | ||
| Fig. 5 Reaction rate of NO in SCR by various alkanes over Cu and Ag catalysts as a function of mean bond energy of linear (solid symbols) and branched (open symbols) alkanes. Reprinted from Appl. Catal., B, 37, J. Shibata, K. Shimizu, A. Satsuma and T. Hattori, Influence of hydrocarbon structure on selective catalytic reduction of NO by hydrocarbons over Cu–Al2O3, pp. 197–204, © 2002, with permission from Elsevier.70 | ||
2.3 Engine bench tests and tolerance to SOx
Some research groups have investigated the possibility of using Ag/Al2O3 catalysts in practical applications for SCR by diesel engine bench tests. Nakatsuji et al.27 have reported a maximum NOx conversion of 75–45% over Ag/Al2O3 in engine bench tests using secondary fuel injection. Kikuchi and Kumagai also examined the use of diesel fuel (a cetane number of 57.9%, sulfur content of 0.005 wt%) as a reducing reagent of SCR over an Ag–Nb/Al2O3 catalyst.26 The NOx conversion of the Ag–Nb/Al2O3 catalyst was around 52% at 773 K, and kept above 50% after the reaction for 53.5 h. Eränen et al. demonstrated an engine bench test using a Volvo 2.5 L diesel engine with injection of Swedish MK1 diesel prior to the 2.5% Ag/Al2O3.28,29 These reports have demonstrated the high feasibility of Ag/Al2O3 catalysts for practical applications. Aardahl et al.72 demonstrated a significant improvement in the SCR activity with Ag/Al2O3 coupled with a plasma fuel reformer in front of the catalytic reactor. In their system, a NOx conversion of around 90% was achieved in the temperature range of 350–500 °C at space velocity of 22 000–28 000 h−1. The promotion effect can be attributed to the on-site formation of oxygenated hydrocarbons in the plasma reactor, which was significant at lower temperatures below 350 °C.Ag/Al2O3 is known to have moderate tolerance to water vapour and SO2.28,29,44 The sulfur tolerance of alumina-based catalysts strongly depends on various reaction conditions.73 The deactivation is mainly caused by the formation of aluminium sulfate, however, the loss of SCR activity due to SO2 is usually only limited to the initial deactivation to some extent and then the stable activity of alumina can be observed at low SO2 level (<100 ppm).74 Although some of the reports have indicated significant deactivation of Ag/Al2O3 in the presence of SO2,58,75,76 the tolerance to SO2, which is strongly affected by Ag loading, reaction temperatures, reductants, additives, support materials, and so on, can be much improved by controlling the reaction atmosphere and appropriate catalyst design. Hickey et al.76 indicated that the use of CeO2–ZrO2 supports confer improved sulfur resistance and activity at lower temperatures compared to alumina-based catalysts. Shimizu et al.77 recently reported that SO2 tolerance is improved by H2 co-feeding into the C3H8-SCR reaction atmosphere as well as an increase in the Ag loading of Ag/Al2O3. The higher SO2 tolerance is caused by the reaction of sulfates on Ag-containing sites with H2, which results in the desorption of SO2 to the gas phase or migration of the sulfates to the alumina surface. He and Yu31 confirmed the stable and sufficient activity in ethanol-SCR on the diesel engine bench test containing SO2. Interestingly, a promotion effect of SO2 on the HC-SCR activity of Ag/Al2O3 can be found in some cases. Angelidis et al.56 reported that the addition of 25–100 ppm of SO2 in the NO–C3H6–C3H8–O2 reaction atmosphere at 480 °C results in an increase in the N2 yield from 22 to 48–55% at the expense of the NO2 yield and hydrocarbon conversions. They assumed that the promotion effect of SO2 on the SCR activity is due to the following reasons; the increased acidity of the sulfated alumina support, the formation of R–SOx species during the reaction, and some modifications of silver species in particle size or oxidation state.
2.4 Formation of by-products
One of the major drawbacks of Ag/Al2O3 catalysts is the formation of harmful by-products, i.e., CO, CH3CHO, and nitrogen-containing molecules, such as NH3, CH3CN, and HCN. Due to the high toxicity of these by-products, several efforts have been made to suppress or decompose them. Miyadera made an attempt to remove by-products produced from ethanol-SCR over Ag/Al2O3, especially at lower temperatures below 350 °C. 60 He found that a combination of other noble metal supported catalysts is effective. These harmful by-products were completely removed by a series of catalysts Ag/Al2O3 + CuSO4/TiO2 + Pt/SiO2 placed separately in the reactor in this sequence.Arve et al. carefully analysed the gas phase by-products in octane-SCR and toluene-SCR over Ag/Al2O3 by GC-MS and detected various types of by-products such as amines, ketones, and cracking products.78 They also demonstrated that a cascade reactor consisting of Ag/Al2O3 and Cu–MFI is very effective for both the increase in NO conversion at lower temperatures and suppression of the formation of these by-products.
Eränen et al.79 noted the production of a considerable amount of CO during the octane-SCR over 2 wt% Ag/Al2O3 catalysts, although the catalyst shows a maximum NO conversion of NO to N2 of approximately 90% at 450 °C and an average NO conversion of 66% in the temperature range of 300–600 °C. Interestingly, the placement of a Pt-based commercial oxidation catalyst after an Ag/Al2O3 catalyst is very effective for the removal of CO from the exhaust by oxidation to CO2, however, almost all activity in terms of NO conversion to N2 was lost by the immediate connection of these catalysts. By extending the distance between the two catalyst beds, the NO conversion was significantly improved. They pointed out the importance of the residence time of the postcatalytic chamber and the contribution of the gas phase reaction after the Ag/Al2O3 catalyst initiated on the catalyst surface.
2.5 Modification of Ag/Al2O3 catalysts
There are not many reports on the modification of Ag/Al2O3 catalysts by additive elements. However, the addition of small amount of precious metals sometimes increases the HC-SCR activity of Ag/Al2O3. Since the insufficient SCR activity of Ag/Al2O3 stems from its low ability to activate hydrocarbons,48,80 one of the roles of additives is the promotion of hydrocarbon activation. The addition of some precious metals, such as Pd and Rh, has been reported to be effective for the promotion of HC-SCR activity of Ag/Al2O3.50,51 Sato et al. found that the addition of small amounts of Rh enhanced the activity of Ag/Al2O3 for the selective reduction of NO with decane at low temperatures.50 Rh-promoted Ag/Al2O3 showed its rather high performance on decane-SCR even in the presence of 1–20 ppm of SO2. Based on the catalytic activity for elementary reactions, it was suggested that the role of added Rh is to enhance the reaction between NOx and decane-derived species, leading to NO reduction.Modification of the support may also be effective for the promotion of HC-SCR activity. In the case of Ag–zeolite catalysts, the acid strength of zeolites plays an important role in controlling the SCR activity. As pointed out by Shibata et al., the acid sites of zeolites stabilize Ag+ ions and partially oxidized Ag species.81–84 The strong support effect of zeolites is also reported by Seijger et al.85 They found that NOx conversion in propane-SCR of silver exchanged zeolites is ranked as Ag–Na-β > Ag–H-ferrierite, Ag–H-β, Ag–K-ferrierite > Ag–H-ZSM-5. Ag–Na-β showed 42% NOx conversion at 300 °C. This may also be applicable to Ag/Al2O3. Shimizu et al.86 recently investigated the effect of various additives (Zn, Mg, Y, P, and Na) to alumina supports of Ag/Al2O3 on C3H8–H2-SCR. Adding Zn and Mg into the alumina support enhanced the conversion of NO to N2, while adding Na and P significantly decreased the NO conversion. Comparing the rate of NO reduction and acid amount determined by NH3-TPD, the contribution of surface acid sites of alumina supports to the promotion of the SCR activity of Ag was suggested. The acid–base property of supports should be the controlling factor for the HC-SCR activity of Ag/Al2O3.
2.6 Addition of H2 into HC-SCR
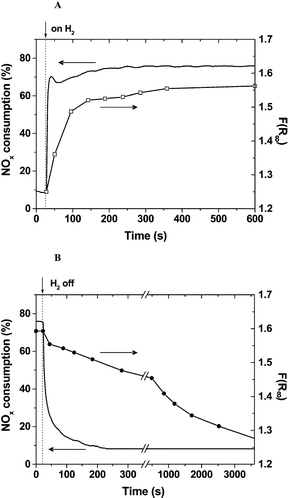
The reaction conditions significantly affect the SCR activity of Ag/Al2O3. The most significant and surprising promotion effect can be obtained by the addition of a small amount of H2. Satokawa, working at the Tokyo Gas Co. Ltd, first reported the promotion effect of H2 on the HC-SCR activity of Ag/Al2O3 at lower temperatures.87,88 As shown in Fig. 6, the addition of 909 ppm of H2 into a C3H8-SCR atmosphere boosted the NO conversion at 673 K from nearly zero to ca. 50%. Interestingly, as shown in Fig. 7, the promotion effect of H2 on NO and propane conversions was reversible depending on the presence of H2 in the gas phase.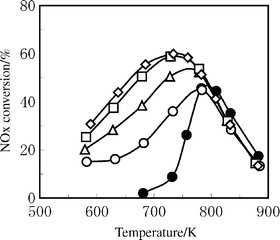 | ||
| Fig. 6 NOx conversion over Ag/Al2O3 at various H2 concentrations. Feed: 91 ppm NO, 91 ppm C3H8, 9.1% O2, 9.1% H2O, and 0 ppm (●), 227 ppm (○), 451 ppm (Δ), 909 ppm (□), or 1818 ppm (◇) H2 in He. GHSV = 44 000 h−1. (Reproduced with permission from ref. 87). | ||
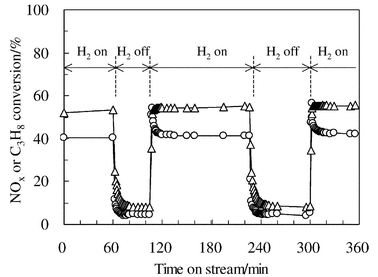 | ||
| Fig. 7 Effect of H2 on the NOx and C3H8 conversions at 673 K as a function of time over Ag/Al2O3. Feed: 91 ppm NO, 91 ppm C3H8, 9.1% O2, 0 or 455 ppm H2, and He as balance. GHSV: 44 000 h−1. Reprinted from Appl. Catal., B, 42, S. Satokawa, J. Shibata, K. Shimizu, A. Satsuma and T. Hattori, Promotion effect of H2 on the low temperature activity of the selective reduction of NO by light hydrocarbons over Ag/Al2O3, pp. 179–184, © 2003, with permission from Elsevier.88 | ||
The promotion effect of H2 was also observed in the SCR with saturated and unsaturated C1–C4 hydrocarbons.87 The “hydrogen effect” was also observed over Ag–zeolite catalysts, such as Ag/MFI and Ag/BEA,81–84 but it was not observed when TiO2, ZrO2, SiO2, or Ga2O3 were used as supports (Table 1).88 Interestingly, H2 does not act as the reducing agent for NO,88,89 but acts as a promoter for the NO reduction by hydrocarbons.88 This remarkable “hydrogen effect” has been confirmed by Burch et al.,90,91 Sazama et al.,92,93 Arve et al.,94 and Richter et al.95
| Catalyst | Temperature/K | NOx conversion to N2 (%) | |
|---|---|---|---|
| With H2 | Without H2 | ||
| a Reprinted from Appl. Catal., B, 42, S. Satokawa, J. Shibata, K. Shimizu, A. Satsuma and T. Hattori, Promotion effect of H2 on the low temperature activity of the selective reduction of NO by light hydrocarbons over Ag/Al2O3, pp. 179–184, © 2003, with permission from Elsevier.88b Feed: 1000 ppm NO, 1000 ppm C3H8, 10% O2, 0 or 5000 ppm H2, and He as balance. Flow rate: 100 cm3 min−1, catalyst: 0.1 g. | |||
| Ag/Al2O3 | 573 | 36 | 0 |
| Ag/TiO2 | 773 | 3 | 3 |
| Ag/ZrO2 | 773 | 4 | 2 |
| Ag/SiO2 | 773 | 0 | 0 |
| Ag/Ga2O3 | 773 | 12 | 34 |
| Co/Al2O3 | 623 | 10 | 12 |
| Pt/Al2O3 | 598 | 6 | 8 |
| Al2O3 | 773 | 35 | 15 |
Since the temperature of diesel exhausts ranges from 473–623 K, a sufficiently high SCR activity at lower temperatures is required for HC-SCR catalysts. The presence of H2 in the exhaust is a very attractive method for boosting the low temperature SCR activity. Burch et al. reported the best SCR performance of Ag/Al2O3 by the use of octane as a reductant of NO.5,90,91Fig. 8 shows NOx conversion of HC-SCR with various hydrocarbons over Ag/Al2O3 in the presence and absence of H2.5 In the absence of H2, the conversion of NO increased from 450 °C for octane-SCR and from 250 °C for butanol- and butanone-SCR. Surprisingly, in the presence of H2, a sufficient NO conversion was observed from 200 °C for these SCR reactions. In particular, in the case of octane-SCR, the NO conversion started from 250 °C, a lower temperature than in the absence of H2, and nearly 100% conversion of NO was achieved in the wide range of temperatures of 200–500 °C.
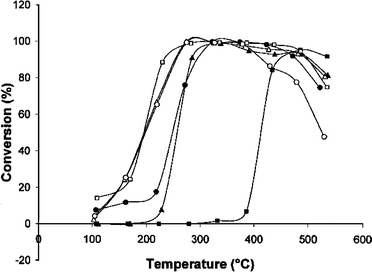 | ||
| Fig. 8 Reduction of NO by various reductants for Ag/Al2O3 catalyst under lean burn conditions with and without the addition of H2. (▲)1-butanol; (Δ) 1-butanol + H2; (●) 2-butanone; (○) 2-butanone + H2; (■) octane; (□) octane + H2.5 | ||
Furthermore, the “hydrogen effect” is also applicable to NH3-SCR over Ag/Al2O3. Richter et al. discovered that above 90% conversion of NO can be achieved in the range of 200–500 °C by the addition of 1 vol% of H2 into NH3-SCR of NO over 5% Ag/Al2O3.96–98 The phenomena is very interesting and may be applied to the exhausts from small sized power plants, rubbish disposal, and so on.
The discovery of a “hydrogen effect” on supported Ag catalysts may provide a novel approach to the design of diesel SCR catalysts. From a scientific point of view, the roles of H2 are still under debate, and various roles of H2 addition have been proposed, i.e., the promotion of hydrocarbon activation to surface oxygenates,80,92 formation of partially charged Agnδ+ cluster,81–84,99 contribution to gas phase radical reactions,100 and generation of peroxide species.101 The details are presented in the section 3.3 and 3.5.
3. Mechanistic investigation of HC-SCR over Ag catalysts
3.1 Role of adsorbed NOx on Ag/Al2O3
There has been much attention on the reaction mechanism of the HC-SCR and active silver species from scientific points of view. The mechanistic investigations have been carried out mainly by means of in situ and operando analysis, especially in situ FTIR. These are mainly focused on: (1) roles of adsorbed NOx species, (2) roles of hydrocarbon-derived surface species in the initial stage of the reaction, and (3) intermediates in the final stage of the reaction.For various types of SCR catalysts, it is generally accepted that gaseous or adsorbed NO2 is the initial intermediate for the selective reduction of NO to N2.102–104 The key roles of nitrates, which are formed from adsorption of gaseous NO2 onto the catalyst surface, have been pointed out in the case of alumina-based catalysts including Ag/Al2O3. We have clarified that the surface nitrates on alumina-based catalysts play a crucial role in the initial stage of the HC-SCR reaction on catalysts such as Al2O3 and Cu–Al2O3 by means of in situ FTIR spectra which are measured under real or pseudo-reaction conditions.104–108 Kinetic analysis of the surface adsorbed species revealed that the selective reaction of NO to N2 initially proceeds by the parallel reactions of the oxidation of NO to surface nitrates (1) and the oxidation of hydrocarbons to surface oxygenates (2), followed by the selective reaction between these species (3). The surface reaction between nitrates and oxygenates leads to the formation of NCO and CN species, and finally leads to the formation of N2 through oxidation or hydrolysis of these nitrogen-containing species, as shown in Fig. 9.
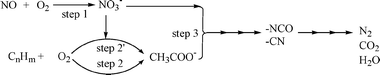 | ||
| Fig. 9 Proposed reaction scheme of the HC-SCR reaction on alumina-based catalysts. | ||
From the in situ FTIR spectra, basically the same reaction pathways were proposed on HC-SCR over Ag/Al2O3.48,49Fig. 10 (0 min) shows the IR spectra of adsorbed species on Ag/Al2O3 prepared by a sol-gel method after exposing the catalyst in a flow of NO + O2 at 623 K. The bands assignable to unidentate nitrate (1554, 1292 cm−1) and bidentate nitrate (1580, 1246 cm−1) and a small band due to weakly adsorbed NO2 (1624 cm−1) are observed. Since the nitrates were not observed in a flow of NO, the formation of nitrates proceeds by NO oxidation to NO2 and the subsequent adsorption of NO2 on basic oxygen sites. Switching to a flow of n-hexane + O2, the nitrate bands gradually disappeared while new bands appeared at around 1570–1580 and 1460 cm−1 assigned to νas(COO) and νs(COO) of adsorbed acetate. In addition, shoulder bands assignable to formate (1378 and 1390 cm−1) and carbonate (1630, 1410, 1300–1336 cm−1) were also observed. Fig. 11 shows a time course of the nitrate concentration in a flow of He, n-hexane or n-hexane + O2. The band intensity was not affected much by a flow of He, indicating thermal desorption of nitrates can be neglected in such a time scale. However, in a flow of n-hexane + O2, the nitrates immediately disappeared within 12 min, indicating that the nitrates reacted with hexane in the presence of O2. The reaction rate of the nitrates can be estimated using the slope of the curve in the figure. The reaction rate of nitrate in a flow of n-hexane + O2 (357 nmol g−1 s−1) was 10 times higher than that of only n-hexane (34 nmol g−1 s−1).
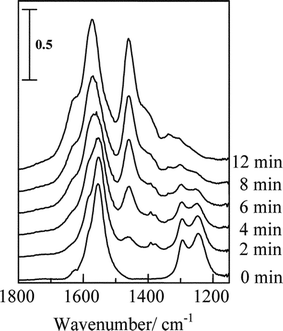 | ||
| Fig. 10 Dynamic changes in the IR spectra as a function of time in flowing n-hexane + O2 on Ag/Al2O3 at 623 K. Before the measurements, the catalyst was pre-exposed to a flow of NO + O2 for 120 min at 623 K. Reprinted from Appl. Catal., B, 30, K. Shimizu, J. Shibata, H. Yoshida and T. Hattori, Silver–alumina catalysts for selective reduction of NO by higher hydrocarbons: structure of active sites and reaction mechanism, pp. 151–162, © 2001, with permission from Elsevier.49 | ||
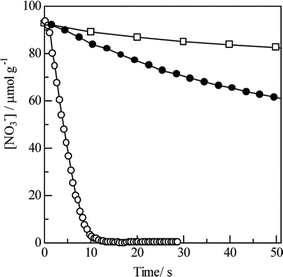 | ||
| Fig. 11 Time dependence of nitrate concentration in flowing He (open square), n-hexane (closed circle) and n-hexane + O2 (open circle) on Ag/Al2O3. Before the measurements, the catalyst was pre-exposed to a flow of NO + O2 for 120 min at 623 K. Reprinted from Appl. Catal., B, 30, K. Shimizu, J. Shibata, H. Yoshida and T. Hattori, Silver–alumina catalysts for selective reduction of NO by higher hydrocarbons: structure of active sites and reaction mechanism, pp. 151–162, © 2001, with permission from Elsevier.49 | ||
These reaction rates of the surface species can be compared with the rates of gas phase products in n-hexane-SCR over Ag/Al2O3, as shown in Fig. 12. In a temperature range of 573–648 K, the initial rates of nitrate consumption were close to the steady state rates of NO reduction, and the apparent activation energy was 61 kJ mol−1, which is comparable to the apparent activation energy for NO reduction (67 kJ mol−1). The good agreement of these rates clearly indicates that N2 is formed via surface nitrates as intermediates. It should be noted that the apparent activation energy for NO reduction increased to 156 kJ mol−1 below 573 K. The apparent activation energy for n-hexane conversion to COx also increased below 573 K. The higher activation energy at lower temperature suggests self-poisoning of nitrate on Ag/Al2O3. Furusawa et al. reported that the ability of Ag/Al2O3 for the oxidation of NO without hydrocarbon reductants was rather lower than that of Ag–MFI.109 They explained that the reaction is retarded on Ag/Al2O3 by the formation of nitrate species, which were produced by the reaction of NO2 with Ag2O phases. Iglesias-Juez et al. carefully investigated the reactivity of nitrates by in situ DRIFTS.110 They elucidated that type II bidentate nitrates (1305 and 1580–1590 cm−1) on Ag/Al2O3 react with C-containing intermediates, forming the potential intermediate for HC-SCR.
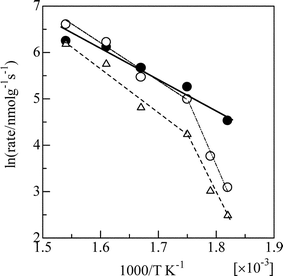 | ||
| Fig. 12 Arrhenius plots of the initial rate of NO3− consumption in n-hexane + O2 (●), and the steady state rates of NO reduction to N2(○) and n-hexane conversion to CO (Δ), for the NO + n-hexane + O2 reaction over Ag/Al2O3. Conditions: NO = 1000 ppm, HC = 6000 ppm C, O2 = 10%. Reprinted from Appl. Catal., B, 30, K. Shimizu, J. Shibata, H. Yoshida and T. Hattori, Silver–alumina catalysts for selective reduction of NO by higher hydrocarbons: structure of active sites and reaction mechanism, pp. 151–162, © 2001, with permission from Elsevier.49 | ||
The crucial role of nitrate species is also clarified by other methods. By means of NO-TPD and NO-TPR spectra of Ag–MFI catalysts, Shi et al.111 proposed that surface nitrate species are formed in NO/O2 and could be effectively reduced by methane to N2. The nitrate species were found to be more reactive to CH4 than O2, being preferentially and selectively reduced to N2. Furthermore, from the temperature-programmed decomposition of AgNO3, they attributed the high activity of Ag–MFI to the formation of strong adsorption centers for surface NO3− species which could be effectively reduced by activated CH4.112
3.2 Role of oxygenated hydrocarbons
As for the hydrocarbon-derived species, a key role of the surface oxygenated hydrocarbons has been pointed out. Our research group has demonstrated that the surface adsorbed acetate species reacts with surface nitrate and leads to the formation of N2 over alumina-based catalysts105–108 including Ag/Al2O3.49Fig. 13 shows the transient behaviour of IR spectra of surface adsorbed species after the NO + n-hexane + O2 reaction followed by treatment in a flow of NO + O2. During the NO + n-hexane + O2 reaction, at 0 min in the figure, bands due to acetate (1460 cm−1), formate (1378 and 1390 cm−1) and unidentate and bidentate nitrates (1292 and 1246 cm−1, respectively) were observed. Shoulder bands are assigned to carbonyl (1720 cm−1) and carbonate species (1630, 1410, 1300–1336 cm−1). In the region of 2300–2000 cm−1, the bands assigned to –NCO bound to Ag+ ions (2232 cm−1) and –CN species (2162 and 2130 cm−1) are observed. After the flowing gas was switched to NO + O2, the bands due to acetate (1460 cm−1) decreased, while nitrate bands progressively appeared. The result can be interpreted as showing that acetate is a reactive species toward nitrates formed by the NO + O2 reaction. The reaction of nitrate with oxygenated hydrocarbon species, possibly acetate, is proposed to be a crucial step in the initial stage of HC-SCR reaction. Furthermore, the band due to Ag+–NCO and CN decreased in a flow of both NO + O2 and O2, indicating the reaction between these species. From the time course of these adspecies, the main reaction pathways of over Ag/Al2O3 catalyst can be depicted as shown in Fig. 9.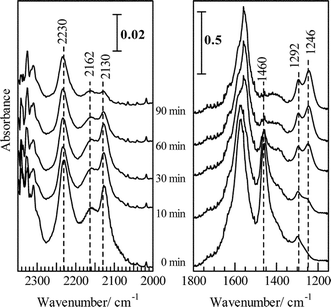 | ||
| Fig. 13 Dynamic changes in the IR spectra as a function of time in a flow of NO + O2 on Ag/Al2O3 at 623 K. Before measurement, the catalyst was pre-exposed to a flow of NO + n-hexane + O2 for 120 min at 623 K. Reprinted from Appl. Catal., B, 30, K. Shimizu, J. Shibata, H. Yoshida and T. Hattori, Silver–alumina catalysts for selective reduction of NO by higher hydrocarbons: structure of active sites and reaction mechanism, pp. 151–162, © 2001, with permission from Elsevier.49 | ||
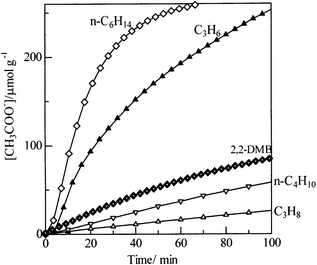
The strong dependence of the rates of NO reduction on the type of hydrocarbon reductants can be rationalized based on the reactivity of the surface oxygenated species.48Fig. 14 shows the dynamic changes in the acetate band formed by the reaction of gaseous hydrocarbons with surface pre-adsorbed nitrate. The response of acetate, i.e., the apparent rate of acetate formation, was in the order of n-hexane > propene > 2,2-dimethylbutane (2,2-DMB) > n-butane > propane. The activity pattern is same as in the reaction rate of the HC-SCR, i.e., the dependence of the rate of NO reduction on the hydrocarbon reductants was as follows: higher alkane (hexane) > lower alkane (propane), alkene (propene) > alkane (propane), linear alkane (hexane) > branched alkane (2,2-DMB). The proposed mechanism in Fig. 9 explains the hydrocarbon effect on the HC-SCR activity: the NO reduction activity depends on the reactivity of the hydrocarbons, which affects the rate of hydrocarbon oxidation to oxygenates and hence the rate of nitrate reaction with oxygenated hydrocarbons.
 | ||
| Fig. 14 Dynamic changes in the IR spectra as a function of time in flowing HC + O2 on Ag/Al2O3 at 623 K. Conditions: HC = 6000 ppc C, O2 = 10%. K. Shimizu, J. Shibata, A. Satsuma and T. Hattori, Phys. Chem. Chem. Phys., 2001, 3, 800. Reproduced by permission of the PCCP Ownership Board.48 | ||
Recently, Yeom et al. identified the intermediates in ethanol-SCR over Ag/Al2O3 by means of FTIR of both gas phase and surface species.113 They observed the formation of ethyl nitrite in the gas phase as an initial intermediate for ethanol-SCR. Fig. 15 shows gas phase FTIR spectra taken after exposing Ag/Al3O3 to various mixed gases. The bands at 1745 and 1671 cm−1 after exposure of Ag/Al3O3 to NO2 + ethanol + O2 were assigned to ethyl nitrite. The shift of the band at 1671 cm−1 (Fig. 15b) to 1642 cm−1 (Fig. 15c) by the feed of 15NO clearly elucidates the formation of N-containing molecules. They confirmed very fast decomposition of ethyl nitrite to acetaldehyde and then acetate from dynamic behaviour. As shown in Fig. 16c, the bands at 1606, 1687, and 1466 cm−1 were observed under ethanol-SCR conditions. The band at 1687 and 1466 cm−1 can be assigned to surface nitrate and acetate, respectively. From the comparison with the spectra of adsorbed nitromethane (Figs. 16f–16g) the shoulder band at 1606 cm−1 under ethanol-SCR conditions (Fig. 16c) was attributed to surface nitromethane. From these detailed results and discussions, they proposed the following reaction scheme for ethanol-SCR
| ethanol → ethyl nitrite → acetaldehyde → acetate → nitromethane → NCO →→ N2. | (2) |
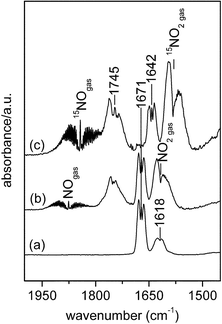 | ||
| Fig. 15 Gas phase FTIR spectra: (a) 2 Torr of ethyl nitrite at 25 °C, (b) spectrum of the gas phase over Ag/γ-Al2O3 after exposure of Ag/γ-Al2O3 to 2 Torr NO2 + 3.8 Torr 2-13C-ethanol + 60 Torr O2, (c) spectrum of the gas phase over Ag/γ-Al2O3 after exposure of Ag/γ-Al2O3 to 2 Torr 15NO + 3.8 Torr ethanol + 60 Torr O2. Prior to admission of the premixture of 3.8 Torr ethanol + 60 Torr O2, the Ag/γ-Al2O3 was pre-exposed to 3.8 Torr of NO2 (or 15NO), © 2006, with permission from Elsevier.113 | ||
 | ||
| Fig. 16 IR spectra of Ag/γ-Al2O3 exposed to: (a) acetic acid, (b) ethanol + O2, (c) ethanol + NO2 + O2, (d) 2-13C ethanol + NO2 + O2, (e) ethanol + O2 + 15NO2, (f) nitromethane and (g) 13C-nitromethane. In each case, the cell was evacuated after 1 min of exposure to the indicated sample. The acetic acid pressure was 0.8 Torr, ethanol partial pressure was 3.8 Torr, NO2 partial pressure was 3.8 Torr, O2 partial pressure was 60 Torr, and nitromethane pressure was 2 Torr. The temperature was 200 °C for traces (a)–(e) and 37 °C for traces (f) and (g), © 2006, with permission from Elsevier.113 | ||
The key role of surface oxygenated hydrocarbons as intermediates for HC-SCR is generally accepted, but the structure of oxygenated hydrocarbons is still a subject of debate, i.e., other molecules are proposed to be possible key species. Iglesias-Juez et al.110 also emphasized the importance of C3H6 activation in C3H6-SCR over Ag/Al2O3, which involves generation of surface carboxylate-type species as a partially oxidized intermediate. In their in situ DRIFTS experiment of Ag/Al2O3 under stepwise treatment by NO, O2, and propene, they observed a group of bands at 1642–1575, 1454–1458, 1378–1392, and 1296–1298 cm−1. They assigned the bands to surface acrylate and postulated that it was of importance in catalytic performance on C3H6-SCR over Ag/Al2O3.
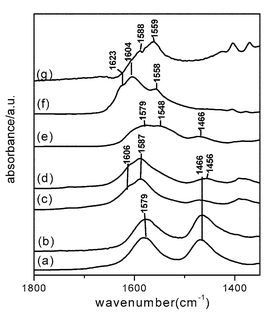
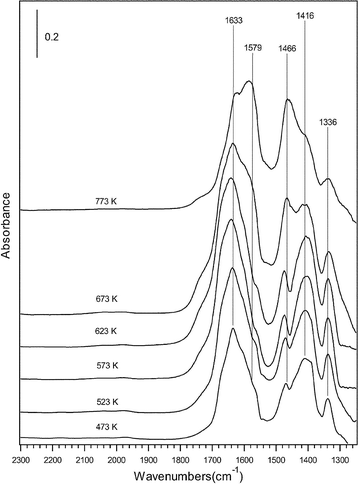
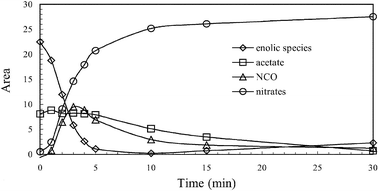
In the case of ethanol-SCR reaction, He et al. proposed that surface enolic species were more reactive adspecies than acetate.31,51,114Fig. 17 shows their in situ DRIFTS spectra of adsorbed species on Ag/Al2O3 during the partial oxidation of EtOH. A series of bands assignable to enolic species were observed at 1633, 1416 and 1336 cm−1 together with the bands due to acetate (νas(OCO) at 1579 cm−1 and νs(OCO) at 1466 cm−1). They tentatively assigned the band at 1633 cm−1 to νas(CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–O−), that at 1416 cm−1 to νs(CH2CH–O−), and that at 1336 cm−1 to C–H deformation mode. As shown in Fig. 18, a sharp decrease in the bands due to enolic species was observed in a flow of NO + O2 on Ag/Al2O3 after the exposure to C2H5OH + O2. The band assignable to enolic species sharply disappeared after 5 min with simultaneous formation of an NCO band. On the other hand, the band assignable to acetate at 1464 cm−1 slowly decreased after 5 min, showing the lower reactivity of acetate than enolic species. From the dynamic changes of these bands in a flow of NO + O2, they concluded that enolic species on Ag/Al2O3 are highly active towards NO2 and NO3−, resulting in the formation of the NCO species which is the key reaction intermediate in the SCR. Although the importance of partial oxidation for HC-SCR on Ag/Al2O3 is commonly indicated in these papers, different types of hydrocarbon-derived intermediates have been proposed. The difference in the assignment of the hydrocarbon-derived intermediates may come from the difference in the reaction conditions, such as the concentration and properties of reductant, catalyst preparation and pretreatment, acidic property of support, and so on.
CH–O−), that at 1416 cm−1 to νs(CH2CH–O−), and that at 1336 cm−1 to C–H deformation mode. As shown in Fig. 18, a sharp decrease in the bands due to enolic species was observed in a flow of NO + O2 on Ag/Al2O3 after the exposure to C2H5OH + O2. The band assignable to enolic species sharply disappeared after 5 min with simultaneous formation of an NCO band. On the other hand, the band assignable to acetate at 1464 cm−1 slowly decreased after 5 min, showing the lower reactivity of acetate than enolic species. From the dynamic changes of these bands in a flow of NO + O2, they concluded that enolic species on Ag/Al2O3 are highly active towards NO2 and NO3−, resulting in the formation of the NCO species which is the key reaction intermediate in the SCR. Although the importance of partial oxidation for HC-SCR on Ag/Al2O3 is commonly indicated in these papers, different types of hydrocarbon-derived intermediates have been proposed. The difference in the assignment of the hydrocarbon-derived intermediates may come from the difference in the reaction conditions, such as the concentration and properties of reductant, catalyst preparation and pretreatment, acidic property of support, and so on.
 | ||
| Fig. 17 In situ DRIFTS spectra of adsorbed species in the steady states on 5 wt% Ag/Al3O3 at different temperatures in a flow of C2H5OH + O2. Conditions: C2H5OH 1565 ppm, O2 10%. Reprinted from Appl. Catal., B, 49, Y. Yu, H. He, Q. Feng, H. Gao and X. Yang, Mechanism of the selective catalytic reduction of NOx by C2H5OH over Ag/Al2O3, pp. 159–171, © 2004, with permission from Elsevier.114 | ||
 | ||
| Fig. 18 Time dependence of the integrated area of the bands in a flow of NO + O2 at 673 K. Before the measurement, the catalysts was pre-exposed to a flow of C2H5OH + O2 for 60 min at 673 K. Conditions: NO 800 ppm, C2H5OH 1565 ppm, O2 10%. Reprinted from Appl. Catal., B, 49, Y. Yu, H. He, Q. Feng, H. Gao and X. Yang, Mechanism of the selective catalytic reduction of NOx by C2H5OH over Ag/Al2O3, pp. 159–171, © 2004, with permission from Elsevier.114 | ||
In the latter part of the HC-SCR reaction, nitrogen-containing species are thought to play a key role in the formation of N2. As already shown in Fig. 13, the bands assignable to cyanide (–CN) and isocyanate (–NCO) can be observed on Ag/Al2O3 under the HC-SCR reaction conditions. Kameoka et al. studied the reactivity of surface isocyanate species with NO, O2 and NO + O2 by pulse reaction of adsorbed NCO species with gaseous O2 or NO + O2.115 After the formation of adsorbed NCO species by exposing Ag/Al2O3 or Al2O3 under ethanol-SCR conditions, they injected several pulses of the oxidants. The adsorbed NCO species on Ag/Al2O3 reacted with O2 or NO + O2 gas to produce N2 effectively at 250 °C. The reactivity of NCO species with the reactant gases was in good agreement with the changes in NCO bands measured by in situ DRIFTS. The results clearly indicate that NCO species is the important intermediate in HC-SCR for the formation of N2. In the case of Al2O3 alone, less N2 was detected in the reaction with NO + O2, indicating that Ag plays an important role in the formation of N2 from NCO.
Bion et al. demonstrated the dynamics of the surface –CN and –NCO species on Ag/Al2O3 under ethanol-SCR by coupling of in situ DRIFT spectroscopy and mass spectrometry.116 In the in situ FTIR spectra, just after the injection of ethanol onto Ag/Al2O3, the characteristic bands of cyanide (–CN) increase in intensity, followed by isocyanates (–NCO). They assigned these bands to silver cyanide (Ag–CN) and its transformation into Al–NCO, respectively. Fig. 19 shows the dynamic changes in the amount of adsorbed NCO species and the gas phase CO2 and NH3 produced during the introduction of H2O pulses on Ag/Al2O3. The behaviour of these species indicates hydrolysis of isocyanate with water vapour into CO2 and NH3. Then, the NH3 thus formed gives rise to N2 through the reaction with NO in excess oxygen.
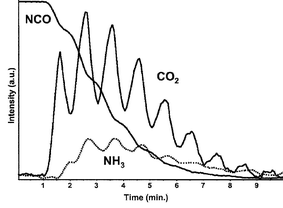 | ||
| Fig. 19 Dynamic changes of NCO consumption and CO2 and NH3 gas species produced during the introduction of H2O pulses on Ag/Al2O3 previously treated under CO + NO at 873 K. Reprinted from J. Catal., 217, N. Bion, J. Saussey, M. Haneda and M. Daturi, Study by in situ FTIR spectroscopy of the SCR of NOx by ethanol on Ag/Al2O3—evidence of the role of isocyanate species, pp. 47–58, © 2003, with permission from Elsevier.116 | ||
The important roles of gas phase reactions are also proposed.99,117,118 Li and Villani emphasized the importance of ammonia formation during NOx trap operation.117 Arve et al. proposed a combination of heterogeneous and homogeneous (radical) reactions over HC-SCR over Ag/Al2O3.100 Radicals of low molecular weight were detected in the gas phase. Cyanogen isocyanate, NC–NCO, was detected and suggested to be a key intermediate in the formation of amines and ammonia via the hydrolysis of isocyanate species. Since these gaseous intermediates significantly influences the HC-SCR performance depending on the reactor design, further clarification of the role of the gaseous active species should be necessary.
3.3 Reaction mechanism of the H2 HC-SCR reaction
The boosting of the HC-SCR activity in the presence of H2 is interpreted as an acceleration of a specific reaction pathway. Satokawa et al. observed the significant promotion of the oxidation reactions of NO to NO2 and propane to COx in the presence of H2.88 The reduction of NO2 by propane, however, was not promoted by the addition of hydrogen. From these results, the promotion of SCR activity by hydrogen was attributed mainly to the promotion of activation of hydrocarbons.Burch et al.90 and Sazama et al.92 also observed the promotion of the activation of octane or decane at lower temperatures. They ruled out the contribution of the enhancement of the oxidation NO to NO2 to the increased rate of the SCR reaction. From transient kinetics and in situ DRIFTS measurements, Burch et al. 90 indicated that hydrogen has a direct role in the reaction mechanism by either promoting the formation and storage of an organic CN species which can then readily reduce NOx and/or removing a species which acts as a poison to the SCR reaction at low temperatures. Sazama et al.92 showed that the reaction steps most enhanced by the addition of hydrogen to the SCR-NOx reaction are the transformations of the intermediate CN species into NCO and oxidation of the hydrocarbon to formates or acrylates.
Shibata et al. clearly demonstrated that the “hydrogen effect” is mainly attributed to the promotion of oxidation of hydrocarbons to surface oxygenates.80 The addition of H2 significantly affected the reaction rates of the surface adsorbed species. Fig. 20 shows the time dependence of the surface concentrations of nitrates and acetate on Ag/Al2O3 during the reaction of surface nitrates with C3H8 + O2 in the absence and presence of H2. From the slope of the transient responses, both the consumption rate of nitrates and the accumulation rate of acetate were greatly enhanced by the addition of H2. The initial rates of accumulation and consumption of surface acetate and nitrates were summarized in Table 2 showing corresponding reaction pathways in Fig. 9. The initial rate of nitrate consumption (run 4) was 89 nmol g−1 s−1 in the presence of H2, while it was below 0.1 nmol g−1 s−1 in the absence of H2. It should be noted that these initial rates are measured at the surface nitrate concentration of 340 μmol g−1 (time = 0 min in the figure), which is higher than that under the steady state (280 μmol g−1) in a flow of NO + C3H8 + O2 + H2. From the estimation of the transient curve at 10 min, the consumption rate of surface nitrates can be determined as 75 nmol g−1 s−1, which corresponds well to the consumption rate of gaseous NO (run 4′, 68 nmol g−1 s−1) in the steady state. The comparison of the surface reaction rates (Table 2) should reflect the “hydrogen effects” observed in the gas phase NO conversion. The formation of acetate by C3H8 oxidation (step 2 and 2′) and nitrates by NO oxidation (step 1) were remarkably promoted by the addition of H2. The acceleration of step 3 by the addition of H2 is caused by increases in the concentration terms of adsorbed species, which is due to the promotion of step 1 and 2′. Since the NO oxidation to nitrates (step 1) is excluded from the rate-determining step in the C3H8-SCR, the promotion of NO oxidation to nitrates by the addition of H2 does not contribute to the promotion of C3H8-SCR. The partial oxidation of C3H8 to mainly acetate at step 2′ is remarkably promoted by H2. This promotion leads to the increase in the production rate of N2via the acceleration of step 3 through an increase in acetate concentration. The appearance of the acetate band by the addition of H2 during the C3H8-SCR is explained by the remarkable promotion of C3H8 oxidation to acetate. From these kinetic analyses, the promotion effect of the addition of H2 on the C3H8-SCR is attributed to the remarkable promotion of partial oxidation of C3H8 to mainly surface acetate.
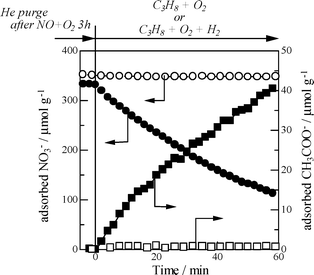 | ||
| Fig. 20 Time dependence of (○, ●) nitrate and (□, ■) acetate concentration in (○, □) C3H8 + O2 and (●,■) C3H8 + O2 + H2 on Ag/Al2O3 at 473 K. Before the measurement, the catalyst was pre-exposed to a flow of NO + O2 for 3 h at 473 K. J. Shibata, K. Shimizu, S. Satokawa, A. Satsuma and T. Hattori, Phys. Chem. Chem. Phys., 2003, 5, 2154. Reproduced by permission of the PCCP Ownership Board.80 | ||
| Run | Gas phase adspecies | Observed change | Step | Rate/nmol g−1 s−1 | |
|---|---|---|---|---|---|
| Without H2 | With H2 | ||||
| a J. Shibata, K. Shimizu, S. Satokawa, A. Satsuma and T. Hattori, Phys. Chem. Chem. Phys., 2003, 5, 2154 – Reproduced by permission of the PCCP Ownership Board.80b C1 basis.c Estimated from slopes at 10 min in Fig. 20.d Not measured. | |||||
| 1 | NO, O2 | Nitrates | 1 | 66 | 560 |
| None | Accumulation | ||||
| 2 | C3H8, O2 | Acetate | 2 | <0.1b | 19b |
| None | Accumulation | ||||
| 3 | NO, O2 | Acetate consumption | −1, −3 | 17b | 350b |
| Acetate | Nitrates accumulation | 1, −3 | 28 | 630 | |
| 4 | C3H8, O2 | Nitrates consumption | −2′, −3 | <0.1 | 89 |
| Nitrates | Acetate accumulation | 2′, −3 | <0.1b | 40b | |
| 4′ | C3H8, O2 | Nitrates consumption | −2′, −3 | —d | 75 |
| Nitratesc | Acetate accumulation | 2′, −3 | —d | 35b | |
| 5 | NO, C3H8, O2 | NO consumption | <0.1 | 68 | |
| Flow reaction | C3H8 consumption | <0.5b | 180b | ||
| 5′ | NO, O2, H2 | NO consumption | <0.1 | ||
| Flow reaction | |||||
3.4 Active Ag species
The importance of the state of Ag species is widely recognized. So far, the HC-SCR activity has been correlated to the specific types of Ag species, such as nano-Ag particles,111 Ag clusters, 50,81–84,99,124 nano-sized Ag2O clusters,119,120 the presence of both Ag+ and Ag particles,111,121,122 and a β silver aluminate-like phase.110,123 The proposed active species on supported Ag catalysts are summarized in Table 3 together with the reaction conditions.| Active Ag Species | Temp/K | Hydrocarbon | Ref. |
|---|---|---|---|
| HC-SCR reaction | |||
| Both Ag+ and metallic Ag | 523–673 | C3H6 | 122 |
| Ag2O cluster | 573–773 | C3H6 | 119, 120 |
| Ag+ compound | 600–750 | C3H6, C2H5OH | 45 |
| Silver aluminate | 600–750 | C3H6 | 110, 123 |
| Ag+ in AgCl | 600–773 | C3H6, C4H8 | 68 |
| Ag+ and Ag nano particle | 673–773 | CH4 | 111, 121 |
| Agn+ cluster | 573–673 | C10H22 | 50, 124 |
| H2-HC-SCR reaction | |||
| Agn+ cluster | 573–723 | C3H8 | 81–84, 99 |
| Ag+ or small Ag2O | 473–673 | C10H22 | 92 |
| Chemical effect | 473–623 | C8H18 | 125 |
| Ag–hydride/peroxide | 473–673 | C10H22 | 101 |
Due to the strong dependence of catalytic performance on Ag loading, Miyadera et al.45 pointed out that the state of the Ag species is the important factor for the HC-SCR activity of Ag/Al2O3. Usually, the highest HC-SCR activity can be observed at Ag loadings of 2–4 wt%. Since these catalysts are white and show no diffraction lines attributable to Ag-derived species in XRD patterns (Ag2O or Ag metal), well-dispersed Ag species are thought to be the active species for the HC-SCR. This idea was also supported by the effect of pre-treatment of Ag/Al2O3.126 Ag/Al2O3 after oxidation in 10% O2 at 873 K showed higher ethanol-SCR and propene-SCR activity than after reduction in H2 at 573 K with metallic Ag particles. From the characterizations by SEM, XRD, and XPS, they concluded that oxidized Ag species interacting with the alumina support were the active phase for the HC-SCR reactions.
Furusawa et al.109,119 examined propylene-SCR in the presence of 5% oxygen over Ag supported on Al2O3, H–MFI and H–Y. Several characterizations revealed that silver was present in the form of Ag2O clusters, but not in metallic form for these catalysts. Comparing the amounts and the size of the Ag2O clusters, the difference in catalytic activity (Ag/Al2O3 > Ag–MFI > Ag–HY) was correlated to Ag2O clusters.
Iglesias-Juez et al.110 suggested that Ag+ species in an alumina matrix are the active species for C3H8-SCR reaction, and correlated the high non-selective hydrocarbon oxidation activity to the formation of metallic Ag. The Ag K-edge XANES and EXAFS data revealed the presence of Ag+ ions in a β-AgAlO2-like phase in 4.5 wt% Ag/Al2O3 that showed the highest activity on C3H8-SCR. They also emphasized the importance of the oxygen lability of the β-AgAlO2-like phase for the formation of surface acrylate from the reduction–oxidation of Ag species.
On the other hand, the formation of Ag clusters is also correlated to the good performance of the SCR reaction in several cases. Shi et al. investigated the effect of silver loading on the structure of silver species as well as on the activity for SCR with methane over Ag/MFI.111 They reported a sigmoid relationship between NO conversion and Ag loading; the NO conversion was increased slightly by the introduction of 3–5 wt% Ag to the zeolite supports but was greatly enhanced on the 7 and 9 wt% Ag/H–MFI catalysts. This result suggests that aggregated silver species are more effective sites for SCR than isolated Ag+ ions, and this was supported by the UV-Vis spectra which showed that the band assignable to nano-sized Ag clusters (410 nm) were observed on the higher Ag loading samples (≥7 wt%). Ag clusters are also identified to be the active species for the propane-SCR in the presence of H2.81–84
Sato et al. reported the strong correlation between the formation of Ag clusters by the addition of promoters and the improvements in the SCR activity.50,124 As shown in Table 4, a considerable promoting effect by Rh addition was found on the catalytic performance of Ag/Al2O3 for decane-SCR of NO at low temperatures. On the other hand, the addition of Ir, Ru, Pd, and Pt decreased the SCR activity of Ag/Al2O3. Fig. 21 shows diffuse reflectance UV-Vis spectra of Ag/Al2O3 and promoted Ag/Al2O3 after subtracting the Al2O3 spectrum. Ag/Al2O3 showed broad absorption bands around 223 and 352 nm, while Ag/Rh/Al2O3 gave an additional broad band around 246 nm, and Ag/Ir/Al2O3 and Ag/Ru/Al2O3 had a broad band around 290 nm. Note that these bands are not attributed to the added Rh, Ir, and Ru. According to their assignments, the bands at 238–272 nm, 275–326 nm, and 330–385 nm are attributed to Agnδ+ clusters, Agn1 small metallic clusters, and Agn2 large metallic clusters, respectively. The band at 223 nm is assigned to the electronic transition from 4d10 to 4d95s1 of highly dispersed Ag+ ions. It is clear from these spectra that Ag+ and Agn2 clusters are the major Ag species on Ag/Al2O3, the band characteristic of Agnδ+ species is observed only on Ag/Rh/Al2O3, while metallic Ag clusters were the main species on Ag/Ir/Al2O3 and Ag/Ru/Al2O3. In situ DRIFT spectra revealed that the formation rate of surface –NCO species, which is known to be a reaction intermediate for the formation of N2, was significantly increased by the addition of Rh to Ag/Al2O3. They concluded that the role of added Rh is to form Agnδ+ clusters, which are more active for the formation of NCO species as a reaction intermediate.
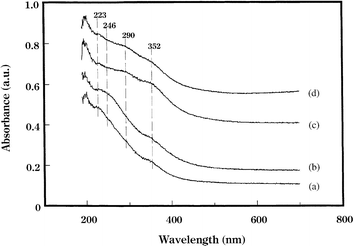 | ||
| Fig. 21 Diffuse reflectance UV-Vis spectra of (a) 4% Ag/Al2O3, (b) 4% Ag/0.05% Rh/Al2O3, (c) 4% Ag/0.05% Ir/Al2O3 and (d) 4% Ag/0.05% Ru/Al2O3. Reprinted from Appl. Catal., B, 44, K. Sato, T. Yoshinari, Y. Kintaichi, M. Haneda and H. Hamada, Remarkable promoting effect of rhodium on the catalytic performance of Ag/Al2O3 for the selective reduction of NO with decane, pp. 67–78, © 2003, with permission from Elsevier.50 | ||
| NO to N2 conversion (%) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Catalyst | 250/°C | 300/°C | 350/°C | 400/°C | 450/°C | 500/°C | 550/°C | 600/°C |
| a Reprinted from Appl. Catal., B, 44, K. Sato, T. Yoshinari, Y. Kintaichi, M. Haneda and H. Hamada, Remarkable promoting effect of rhodium on the catalytic performance of Ag/Al2O3 for the selective reduction of NO with decane, pp. 67–78, © 2003, with permission from Elsevier.50b Reaction conditions: 1000 ppm NO, 10% O2, 333 ppm n-C10H22, 10% H2O, He balance, W/F = 0.05 g s cm−3. | ||||||||
| 4% Ag/Al2O3 | 13 | 30 | 22 | 12 | 10 | 15 | 13 | 12 |
| 4% Ag/0.05% Rh/Al2O3 | 22 | 47 | 40 | 29 | 12 | 6 | 4 | 3 |
| 4% Ag/0.05% Ir/Al2O3 | 6 | 3 | 5 | 10 | 6 | 4 | 3 | 3 |
| 4% Ag/0.05% Ru/Al2O3 | 27 | 10 | 6 | 3 | 2 | 2 | 4 | 3 |
| 4% Ag/0.05% Pd/Al2O3 | 1 | 4 | 4 | 3 | 3 | 2 | 2 | 4 |
| 4% Ag/0.05% Pt/Al2O3 | 2 | 4 | 4 | 3 | 1 | 1 | 0 | 0 |
It can be seen that the proposed active Ag species summarized in Table 3 depends on the reaction conditions, especially on the type of reductant and the reaction temperatures. In the case of SCR by alkene, Ag+ ions or Ag+ containing compounds have been proposed as active species. In the case of the SCR with alkane, which is carried out at higher temperatures because of the lower reactivity of alkane than alkene, Ag clusters are mainly proposed as the active species. These correlations can be roughly rationalized by assuming (1) the activation of hydrocarbon reductants is the critical step for HC-SCR and (2) agglomerated Ag species such as Ag clusters are more active for hydrocarbon activation than dispersed Ag species such as Ag+ ion and silver aluminate. However, we should take high mobility of supported Ag species 127–129 into account when determining the active Ag species. For example, the reversible agglomeration–dispersion behaviour of silica supported Ag species can be observed during CO oxidation. Qu et al.130 investigated restructuring and re-dispersion of Ag on SiO2 under the oxygen–hydrogen pre-treatment. Oxygen pre-treatment at 773 K results in the formation of subsurface oxygen and also activates the catalyst for CO oxidation. The subsequent He treatment at lower temperatures (373–573 K) causes surface faceting and re-dispersion of the surface Ag particles. Interestingly, the re-dispersion occurs without destroying the silver subsurface species. Prolonged H2 treatment at above 573 K includes the aggregation of silver particles. The effect of the oxidation/reduction cycle is reversible. Similar phenomena are expected on supported Ag catalysts under the HC-SCR reaction conditions. Therefore, careful in situ characterizations should be necessary for the determination of active Ag species for the HC-SCR.
3.5 Active species in presence of hydrogen
The “hydrogen effect” on HC-SCR over supported Ag catalysts has been confirmed by several research groups, although the reason for the enhancement of the SCR activity is still unclear. Modification of surface Ag species by H2 is one possible reason. Our research group has found that the promotion effect by H2 on HC-SCR activity can be attributed to the formation of Ag clusters by means of a in situ UV-Vis spectrophotometer equipped with an in situ flow cell (JASCO UHR-630).99Fig. 22 shows in situ UV-Vis spectra of 2 wt% Ag/Al2O3 when flowing various model feed gases at 523 K. In a flow of O2, a band assignable to the 4d10 to 4d9s1 transition band of the Ag+ ion 50,131–136 was observed below 300 nm. In the presence of H2, the spectra showed other broad bands at 300–400 nm, which can be assigned to Ag clusters including ionic Agnδ+ clusters and small metallic Agn clusters.50,137–141 In a flow of NO + O2, these bands disappeared again and the bands due to Ag+ ions was observed, indicating Ag clusters reversibly dispersed in the oxidative atmosphere. The formation of Ag clusters is coincident with the C3H8-SCR activity shown in Fig. 7. The NO conversion was negligible in the absence of H2, while the NO conversion was boosted up to 40% in the presence of H2, and the SCR activity again became negligible after an evacuation of H2. The reversible changes in the Ag species on alumina indicate that a possible reason for the promotion effect by H2is the formation of Ag clusters. Interestingly, it was found from the time course of the band of Ag clusters at 306 nm that the reversible formation of Ag clusters is a very rapid process, as shown in Fig. 23. The formation of Ag clusters in H2 + O2 was complete within 30 s, while re-dispersion into isolated Ag+ ions in NO + O2 takes approximately 1 min. The rates of formation and dispersion of Ag clusters are strongly dependent on the Ag content. Since Ag clusters and Ag metal particles are reported to be highly active for oxidation of hydrocarbons and NO,44,131,132,142 the transformation of Ag species can reasonably explain the changes in the activity in surface reaction steps. The reversible transformation of Ag species between Ag+ ions to Ag clusters and Ag metal by H2 at relatively low temperatures is a well-known phenomenon on Ag–zeolites.127–129 However, it is interesting that the formation of Ag clusters was observed under highly oxidized atmospheres, i.e., in the presence of NO and excess oxygen. The results of in situ UV-Vis spectra indicate the contribution of the following reversible changes of Ag species to the “hydrogen effect” on HC-SCR . | (3) |
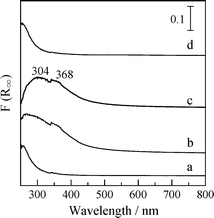 | ||
| Fig. 22 In situ UV-Vis spectra of 2 wt% Ag/Al2O3 at 523 K in a flow of (a) 10% O2, (b) 10% O2 + 0.5% H2, (c) difference spectrum of (b) and (a), and (d) 0.1% NO + 10% O2. Reprinted from Stud. Surf. Sci. Catal., 145, A. Satsuma, J. Shibata, A. Wada, Y. Shinozaki and T. Hattori, In-situ UV-visible spectroscopic study for dynamic analysis of silver catalyst, pp. 235-238, © 2003, with permission from Elsevier.99 | ||
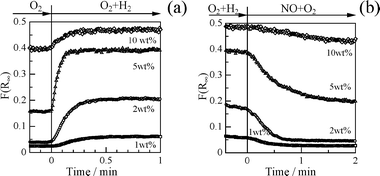 | ||
| Fig. 23 Time course of the band at 306 nm at 523 K under a flow of (a) 10% O2 to 10% O2 + 0.5% H2 and (b) O2 + H2 mixture to 0.1% NO + 10% O2 mixture. Reprinted from Stud. Surf. Sci. Catal., 145, A. Satsuma, J. Shibata, A. Wada, Y. Shinozaki and T. Hattori, In-situ UV-visible Spectroscopic Study for Dynamic Analysis of Silver Catalyst, pp. 235-238, © 2003, with permission from Elsevier.99 | ||
Richter et al. also confirmed the reversible reduction/re-oxidation of nano-sized Ag2O clusters on Ag/Al2O3 to intermediate Ag0 on a short-term scale by means of TEM, TPR, and DTA analysis under the periodic treatment by H2 and O2.95 Since more oxidized adspecies (nitrite/nitrate) are observed in the presence of H2, they concluded that the hydrogen effect can be attributed to a dissociative activation of gas phase oxygen on Ag0 which leads to conversions of gaseous NO to adsorbed nitrate and propane to acetates and other oxygenates. It should be noted that the hydrogen effect was not limited to Ag/Al2O3 prepared by impregnation but also to that prepared by sol-gel processes starting from colloidal alumina sources.
The formation of Ag clusters in the presence of H2 is more clearly observed in Ag–zeolites. Ag–MFI and Ag–BEA also show boosted SCR activity in the presence of H2, as reported by our group.84 In the absence of H2, the reaction rate of NO for C3H8-SCR at 573 K was less than 50 nmol g−1 s−1 for Ag–MFI and Ag–BEA at an Ag/Al ratio of approximately 0.6. In the presence of H2, the reaction rate of NO significantly increased to 360 nmol g−1 s−1 for Ag–MFI (Ag/Al = 0.57) and 130 nmol g−1 s−1 for Ag–BEA (Ag/Al = 0.62). On the other hand, the “hydrogen effect” was not observed for Ag–Y and Ag–MOR. It should be noted that, in the case of Ag–MFI, the reaction rate of NO significantly increased with an increase in the Ag : Al ratio, indicating that the ratio of Ag and amount of acid sites is one of the controlling factors for the promotion effect of H2. The strong dependence of the “hydrogen effect” on the type of acid sites suggests another property of acid sites, i.e., acid strength, is also the controlling factor. Furthermore, the sigmoid relationship was observed between the Ag : Al ratio and the reaction rate of NO of Ag–MFI. Richter et al. also found the similar relation in H2 C3H8-SCR over Ag/Al2O3.95 The “hydrogen effect” is very significant on 1–2 wt% Ag/Al2O3 but not in 0.5 wt% Ag/Al2O3. These results suggests that the addition of hydrogen is not effective for highly dispersed Ag species but effective for moderately dispersed Ag species, i.e., the structure of Ag species is the important factor for the “hydrogen effect”.
Fig. 24 shows UV-Vis spectra of Ag–zeolites in the absence and presence of 0.5% H2. In the presence of H2, the bands above 260 nm indicate the formation of Ag clusters due to partial reduction and agglomeration of Ag species, however, the type of Ag clusters is strongly affected by the zeolite supports. An adsorption band due to the Ag+ ion was observed below 230 nm after C3H8-SCR in the absence of H2. After C3H8-SCR in the presence of 0.5% H2, the spectrum of Ag-MOR was hardly changed, while new bands appeared at 260 and 285 nm due to the Agnδ+ cluster for Ag–MFI. The longer wavelength of the bands of Ag–BEA indicates the formation of larger sized Ag clusters than on Ag–MFI. The extent of the increase in NO conversion by the “hydrogen effect”, inserted in the figure, is in accordance with the formation of Agnδ+ clusters, i.e., the “hydrogen effect” was most significant on Ag–MFI and was not observed on Ag–MOR. Therefore, it is concluded that moderately agglomerated Agnδ+ clusters are highly active species for the C3H8-SCR in the presence of H2, and that the role of H2 is to reduce Ag+ ions to Agnδ+ clusters. Interestingly, the stability of charged Ag species (Ag+ and Agnδ+ clusters) under the H2 HC-SCR (Ag–MOR > Ag–MFI > Ag–BEA) was well correlated to the acid strength of the zeolites, i.e., the heat of adsorption of NH3 on H-form zeolites is in the order MOR (145 kJ mol−1) > MFI (130 kJ mol−1) > BEA (120 kJ mol−1).143–145 It can be rationalized that the stronger acid sites, i.e., the ion-exchange sites of zeolite, stabilize the positively charged Ag+ ions or Agnδ+ clusters, while weaker acid sites easily release Ag+ to form partially reduced Ag species by reduction with H2.
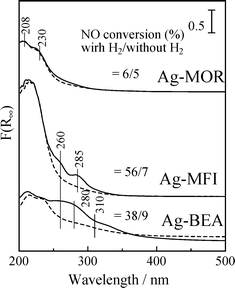 | ||
| Fig. 24 UV-Vis spectra of Ag–MFI, Ag–BEA, Ag–MOR and Ag–Y after (dotted line) C3H8-SCR and (solid line) C3H8-SCR with 0.5% H2 at 573 K. Conditions: NO = 0.1%, C3H8 = 0.1% and O2 = 10%. | ||
Comparing Fig. 22 and 24, Ag–zeolites give sharper UV-Vis bands after H2 HC-SCR than Ag/Al2O3. The broader UV-Vis spectra of Ag/Al2O3 reflect a wide distribution of size or charge of Ag species, which may be due to heterogeneous surface properties of alumina. The ordered structure and uniform chemical profiles of zeolites may result in the formation of uniform Ag species, i.e., Ag–zeolite catalysts would be good models for the supported Ag catalysts for HC-SCR. Detailed structures were characterized for the Agnδ+ clusters on Ag–MFI.82 The H2-TPR result suggests that the average charge of the supported Ag species is +0.5 in the temperature range 373–573 K. Combined with the UV-Vis bands at 255 and 305 nm and the Ag K-edge EXAFS result, the average structure of the clusters is identified as Ag42+. UV-Vis and Ag K-edge XANES/EXAFS results reveals that, during C3H8-SCR with H2 at 573 K, part of the Ag+ ions are converted to Agnδ+ clusters, whose average structure can be close to Ag42+.
Recently, Sazama et al. investigated the dynamic analysis of the formation and re-dispersion of Ag clusters on alumina under realistic reaction conditions by in situ UV-Vis spectra using a spectrometer built in house.92 The bands at 41 600 cm−1 (240 nm) and 46 600 cm−1 (215 nm) attributable to isolated Ag+ ions were observed in in situ UV-Vis spectra under reaction condition of decane-SCR-NO without H2, and the additional band at around 31 000 cm−1 (323 nm) assignable to Agnδ+ clusters was also observed in the presence of H2. The absorption band observed for Ag/Al2O3 was attributed to Ag8δ+ clusters having cubic symmetry.139 The preferable formation of rather unstable Ag4δ+ clusters in MFI may be due to the steric hindrance of the zeolite channel. Fig. 25 shows the response of NO conversion and the intensity of the UV-Vis band at 31 000 cm−1 (323 nm) assignable to the Ag cluster during time-resolved experiments with hydrogen switching on/off in the decane-SCR at 523 K. The NOx consumption increased sharply within seconds after the addition of H2, with a sharp increase in NOx conversion. After hydrogen was switched off, NOx conversion decreased with a slightly longer response (within 4 min). Comparing the responses of the Agnδ+ cluster and NO conversion, the dispersion of Ag clusters to Ag ions was slower than the decrease in NO conversion. Their group also examined the effect of co-feeding H2 and CO into the HC-SCR by using in situ UV-Vis.146 Although only the addition of H2 enhanced decane-SCR, the formation of Agnδ+ clusters was observed in the treatment with both H2 and CO. From this, they claimed that the formation of Ag clusters is not essential for the enhancement of the SCR activity in the presence of H2, and that another effect, such as contributions from hydroperoxy and hydroxy radical species, is more important for the HC-SCR activity in the presence of H2.
 | ||
| Fig. 25 Effect of hydrogen switching on and off on decane-SCR-NO over Ag/Al2O3 at 523 K, 0.1% NO, 0.06% decane, 6% O2 and 0 or 0.2% H2. GHSV = 60 000 h−1. NOx consumption and intensity of the UV-Vis band at 31 000 cm−1 (323 nm) as a function of time and hydrogen switching (A) on and (B) off. Reprinted from J. Catal., 232, P. Sazama, L. Čapek, H. Drobná, Z. Sobalík, J. Dědeček, K Arve and B. Wichterlová, Enhancement of decane-SCR-NOx over Ag/alumina by hydrogen. Reaction kinetics and in situ FTIR and UV–vis study, pp. 302–317, © 2005, with permission from Elsevier.92 | ||
Breen et al.125 applied in situ EXAFS for the determination of Ag species of Ag/Al2O3 under the H2–HC-SCR conditions. In their EXAFS spectra, the change of Ag structure by the presence of H2 was not significant. The Agnδ+ (n = ca. 3 from the coordination number) cluster was already present on the alumina support under SCR conditions without H2. Furthermore, the Agnδ+ cluster was also observed in the presence of CO, although a promoting effect was not observed in the CO–HC-SCR. They concluded that the enhanced activity in the presence of hydrogen is thought to be due to a chemical effect and not the result of a change in the structure of the active sites.
One may wonder what the chemical effect on HC-SCR in the presence of H2 is. Recently, Sazama et al. published very interesting communication.101Table 5 shows the effect of hydrogen and hydrogen peroxide on the C10H22-SCR over Ag/Al2O3 at 473 K.101 The conversion of NO is negligible without co-feeding hydrogen or hydrogen peroxide. The NO conversion significantly increased to 60% in the presence of 0.2% H2, and very interestingly, the promotion effect was also observed in decane-SCR with 0.2% of hydrogen peroxide as well as the “hydrogen effect”. At the same time, decane conversion also increased from nearly zero to 12%. They attributed this promotion effect of SCR by hydrogen peroxide to highly reactive hydroxy and hydroperoxy radicals formed from hydrogen peroxide. It should be noted that in the decane-SCR with hydrogen peroxide, the NO2 yield was higher than that with H2, which may be due to the reaction between hydroxy radicals, HO2 + NO → NO2 + OH. The similarity of the effect of H2 and hydrogen peroxide on decane-SCR performance strongly suggests the role of hydroperoxy-like species during HC-SCR in the presence of H2.
Very recently, Kondratenko et al.147 reported an advanced understanding of the promotion effect of H2 on the NH3-SCR reaction based on the results of a transient low-pressure technique, the temporal analysis of products (TAP) reactor, in combination with isotopic traces. They found that the NH3-SCR activity increased tremendously after ex situ reduction of Ag/Al2O3 in a hydrogen flow at 373 K for 30 min. This observation was related to the creation of reduced Ag species, which catalyze O2 and NO dissociation. The obtained adsorbed oxygen species plays a key role in NH3 dehydrogenation, yielding reactive intermediates for N2 formation. They clearly demonstrated that the reduction of Ag surface is an essential requirement for the promotion effect of H2.
Given the fact that the presence of H2 significantly promotes the activation of hydrocarbons, it is reasonable to consider that the most important role of hydrogen is the formation of reactive oxygen species involved in the oxidative activation of hydrocarbons. It is known that the addition of hydrogen also promotes the partial oxidation of hydrocarbons such as CH4,148–150 ethane,151 C3H6,152 and benzene.153 Wang et al.149,151 investigated the promotion effect of hydrogen on the selective oxidation of methane or ethane by oxygen over an iron phosphate catalyst, and found the formation of peroxide (O22−) on the catalyst. They proposed that hydrogen, acting as the electron donor as well as the proton donor, reductively activates molecular oxygen. It is reasonable to extend these roles of active oxygen to the H2-HC-SCR over supported Ag catalysts. Furthermore, in the case of the H2-HC-SCR over supported Ag catalysts, the dependence of NO conversion on Ag content and UV-Vis spectra suggested an essential contribution of Ag cluster.81–84 The lesser “hydrogen effect” on the Ag catalysts of low Ag content,81,82,112,154 indicates the isolated Ag+ is not effective even if H2 is present in reaction atmospheres. Summarizing all the related data, it can be concluded that both the formation of Ag clusters and hydroperoxy-like species are essential for the “hydrogen effect”. The contribution of these factors on the “hydrogen effect” can be rationalized by considering the following close relationship between Ag clusters and Ag–hydride.155–157 Spectroscopic evidence on the interaction of hydrogen with the partially reduced Ag3+ cluster in Ag-A zeolite has been well investigated using 1H MAS NMR.157 Hydrogen dissociates over Ag3+ sites to generate an acidic proton and Ag3–H. It is reasonable to assume that hydrogen addition results in the formation of hydride on silver clusters, which may react with oxygen to form a reactive oxidant such as hydroperoxy radicals (HO2), peroxide (O22−) or superoxide ions (O2−).
Conclusions
Ag/Al2O3 is a promising candidate for practical uses of selective catalytic reduction of NO by hydrocarbons (HC-SCR). Several engine bench tests demonstrated the high catalytic performance of Ag/Al2O3 and its tolerance to SOx and water vapour. Due to the high sensitivity of HC-SCR activity of supported Ag catalysts to the reaction conditions, the control of the reaction conditions by engine operation and secondary fuel injection would be the key factor for creating effective deNOx in practical automotive engines. The investigations on the reaction mechanism clarified the crucial role of nitrate formed from NO2 and oxygenated hydrocarbons formed from the partial oxidation of hydrocarbon reductants. The SCR reaction proceeds by the surface reaction between nitrate and oxygenated hydrocarbons, such as acetate, and N2 is produced via nitrogen-containing intermediates, i.e., CN, NCO and NH3. The oxidative activation of hydrocarbons is the key step for obtaining high SCR activity at lower temperatures. The addition of hydrogen is effective for enhancing the low temperature SCR activity. Although the mechanism of the “hydrogen effect” is under debate, the key roles of both the formation of Ag clusters and hydroperoxy-like species have been highlighted.Acknowledgements
A part of this study was supported by the Grant-in-Aid from the Ministry of Education, Science, Sports and Culture in Japan. The authors are deeply indebted to co-authors of the papers cited in this Invited Article.References
- J. N. Armor, Appl. Catal., B, 1992, 1, 221 CrossRef CAS
.
- http://unfccc.int/resource/docs/convkp/kpeng.html.
- http://www.mizuho-ir.co.jp/english/knowledge/wtwghg041130.html.
- H. S. Gandhi, G. W. Graham and R. W. McCabe, J. Catal., 2003, 216, 433 CrossRef CAS
- R. Burch, Catal. Rev. Sci. Eng., 2004, 46, 271 CrossRef CAS
- S. Matsumoto, Catal. Today, 2004, 90, 183 CrossRef CAS
- F. Klingstedt, K. Arve, K. Eränen and D. Y. Murzin, Acc. Chem. Res. DOI:10.1051/ar050185k
- J. Suzuki and S. Matsumoto, Top. Catal., 2004, 28, 171 CrossRef CAS
- T. Mizuno, N. Takagi, S. Ohkawara, K. Senda and K. Inuzuka, Shokubai (Catalyst), 2004, 46, 174 Search PubMed
- http://www.toyota-caribbean.com/technology/engines/dcat.html.
- N. Takahashi, H. Shinjoh, T. Iijima, T. Suzuki, K. Yamazaki, K. Yokota, H. Suzuki, N. Miyoshi, S. Matsumoto, T. Tanizawa, T. Tanaka, S. Tateishi and K. Kasahara, Catal. Today, 1996, 27, 63 CrossRef CAS
- S. Matsumoto, Catal. Today, 1996, 29, 43 CrossRef CAS
- M. Koebel, M. Elsener and M. Kleemann, Catal. Today, 2000, 59, 335 CrossRef CAS
- J. H. Baik, S. D. Yim, I.-S. Nam, Y. S. Mok, J.-H. Lee, B. K. Cho and Se H. Oh, Top. Catal., 2004, 30/31, 37 CrossRef
- N. Shirahama, I. Mochida, Y. Korai, K.-H. Choi, T. Enjoji, T. Shimohara and S. Yasutake, Appl. Catal., B, 2004, 57, 235
- J. A. Sullivan and O. Keane, Appl. Catal., B, 2005, 61, 244 CrossRef CAS
- http://www.daimlerchrysler.com/.
- http://www.nissandiesel.co.jp/lineup/quon/index.html.
- M. Iwamoto, H. Yahiro, Y. Yu-u, S. Shundo and N. Mizuno, Shokubai (Catalyst), 1990, 32, 430 Search PubMed
- W. Held, A. König, T. Richter and L. Pupper, SAE Technical Paper Series, 1990, 900496 Search PubMed
- M. Iwamoto and H. Hamada, Catal. Today, 1991, 10, 57 CrossRef CAS
- M. Iwamoto and H. Yahiro, Catal. Today, 1994, 22, 5 CrossRef CAS
- M. Iwamoto, Catal. Today, 1996, 29, 29 CrossRef CAS
- Y. Traa, B. Burger and J. Weitkamp, Microporous Mesoporous Mater., 1993, 30, 3
- M. Iwamoto, Stud. Surf. Sci. Catal., 2000, 130, 23
- T. Kikuchi and M. Kumagai, Sekiyu Gakkaishi, 1998, 41, 173 Search PubMed
- T. Nakatsuji, R. Yasukawa, K. Tabata, K. Ueda and M. Niwa, Appl. Catal., B, 1998, 17, 333 CrossRef CAS
- K. Eränen, L.-E. Lindfors, A. Niemi, P. Elfving and L. Cider, SAE Technical Paper Series, 2000-01-2813 Search PubMed.
- L.-E. Lindfors, K. Eranen, F. Klingstedt and D. Yu. Murzin, Top. Catal., 2004, 28, 185 CrossRef CAS
- J. F. Thomas, S. A. Lewis, G. B. Bunting, J. M. Storey, R. L. Graves and P. W. Park, SAE Technical Paper Series, 2005-01-1082 Search PubMed.
- H. He and Y. Yu, Catal. Today, 2005, 100, 37 CrossRef CAS
- M. Iwamoto and T. Zengyo, Chem. Lett., 1997, 1283 CrossRef CAS
- M. Iwamoto, T. Zengyo, A. M. Hernandez and H. Araki, Appl. Catal., B, 1998, 17, 259 CrossRef CAS
- S. Yoon, A. G. Panov, R. G. Tonkyn, A. C. Ebeling, S. E. Barlow and M. L. Balmer, Catal. Today, 2002, 72, 243–251 CrossRef CAS
- H. Miessner, K.-P. Francke and R. Rudolph, Appl. Catal., B, 2002, 36, 53 CrossRef CAS
- H. Miessner, K.-P. Francke and R. Rudolph, Th. Hammer, Catal. Today, 2002, 75, 325 CrossRef CAS
- R. G. Tonkyn, S. E. Barlow and J. W. Hoard, Appl. Catal., B, 2003, 40, 207 CrossRef CAS
- B. Wen, Y. H. Yeom, E. Weitz and W. M. H. Sachtler, Appl. Catal., B, 2004, 48, 125 CrossRef CAS
- M. Li, J. Henao, Y. Yeom, E. Weitz and W. M. H. Sachtler, Catal. Lett., 2004, 98, 5 CrossRef CAS
- J. H. Kwak, J. Szanyi and C. H. F. Peden, Catal. Today, 2004, 89, 135 CrossRef CAS
- D. N. Tran, C. L. Aardahl, K. G. Rappe, P. W. Park and C. L. Boyer, Appl. Catal., B, 2004, 48, 155 CrossRef CAS
- K. G. Rappe, J. W. Hoardb, C. L. Aardahla, P. W. Parkc, C. H. F. Pedena and D. N. Trana, Catal. Today, 2004, 89, 143 CrossRef CAS
- A. P. Kozlova, H. Y. Law, M. C. Kung and H. H. Kung, Catal. Lett., 2004, 95, 211 CrossRef CAS
- T. Miyadera and K. Yoshida, Chem. Lett., 1993, 1483 CAS
- T. Miyadera, Appl. Catal., B, 1993, 2, 199 CrossRef CAS
- K. Shimizu, J. Shibata, A. Satsuma and T. Hattori, Chem. Lett., 1999, 1079 CrossRef CAS
- K. Shimizu, A. Satsuma and T. Hattori, Appl. Catal., B, 2000, 25, 239 CrossRef CAS
- K. Shimizu, J. Shibata, A. Satsuma and T. Hattori, Phys. Chem. Chem. Phys., 2001, 3, 880 RSC
- K. Shimizu, J. Shibata, H. Yoshida, A. Satsuma and T. Hattori, Appl. Catal., B, 2001, 30, 151 CrossRef CAS
- K. Sato, T. Yoshinari, Y. Kintaichi, M. Haneda and H. Hamada, Appl. Catal., B, 2003, 44, 67 CrossRef CAS
- H. He, J. Wang, Q. Feng, Y. Yu and K. Yoshida, Appl. Catal., B, 2003, 46, 365 CrossRef CAS
- S. Sumiya, M. Saito, H. He, Q. C. Feng, N. Takezawa and K. Yoshida, Catal. Lett., 1998, 50, 87 CrossRef CAS
- A. Abe, N. Aoyama, S. Sumiya, N. Kakuta and K. Yoshida, Catal. Lett., 1998, 51, 5 CrossRef CAS
- F. C. Meunier and J. R. H. Ross, Appl. Catal., B, 2000, 24, 23 CrossRef CAS
- S. Satokawa, K. Yamaseki and H. Uchida, Appl. Catal., B, 2001, 34, 299 CrossRef CAS
- T. N. Angelidis, S. Christoforou, A. Bongiovanni and N. Kruse, Appl. Catal., B, 2002, 39, 197 CrossRef CAS
- P. W. Park and C. L. Boyer, Appl. Catal., B, 2005, 59, 27 CrossRef CAS
- V. Houel, D. James, P. Millington, S. Pollington, S. Poulston, R. Rajaram and R. Torbati, J. Catal., 2005, 230, 150 CrossRef CAS
- T. Miyadera, Appl. Catal., B, 1997, 13, 157 CrossRef CAS
- T. Miyadera, Appl. Catal., B, 1998, 16, 155 CrossRef CAS
- J. S. Yoo, M. J. Kha, S. J. Lee, J. K. Lee and D. S. Kim, Extended Abstract of 10th Japan-Korea Symposium on Catalysis, Matsue, Japan, 2005, 101 Search PubMed
- F. Witzel, G. A. Still and W. K. Hall, J. Catal., 1994, 149, 229 CrossRef CAS
- B. H. Engler, J. Leyer, E. S. Lox and K. Ostgathe, Stud. Surf. Sci. Catal., 1996, 96, 529
- R. Burch and P. J. Millington, Catal. Today, 1995, 26, 185 CrossRef CAS
- R. Burch and D. Ottery, Appl. Catal., B, 1997, 13, 105 CrossRef CAS
- R. Burch, P. Fornasiero and B. W. L. Southward, J. Catal., 1999, 182, 234 CrossRef CAS
- G. Delahay, E. Ensuque, B. Coq and F. Figueras, J. Catal., 1998, 175, 7 CrossRef CAS
- E. F. Iliopoulou, A. P. Evdou, A. A. Lemonidou and I. A. Vasalos, Appl. Catal., A, 2004, 274, 179 CrossRef CAS
- B. Wicherlova, Top. Catal., 2004, 28, 131 CrossRef
- J. Shibata, K. Shimizu, A. Satsuma and T. Hattori, Appl. Catal., B, 2002, 37, 197 CrossRef CAS
- B. K. Hodnett, Hetetogeneous Catalytic Oxidation, John Wiley and Sons, Chichester, 2000, p. 82 Search PubMed
- C. L. Aardahl, R. T. Rozmiarek, K. G. Rappe, D. P. Mendoza and P. W. Park, Abst. 13th Int. Congr. Catal., 2004, O6–011 Search PubMed
- R. Burch, J. P. Breen and F. C. Meunier, Appl. Catal., B, 2002, 39, 283 CrossRef CAS
- M. Tabata, H. Tsuchida, K. Miyamoto, T. Yoshinari, H. Yamazaki, Y. Kintaichi, M. Sasaki, T. Ito and H. Hamada, Appl. Catal., B, 1995, 6, 169 CrossRef CAS
- K. Nagashima, M. Nagata, K. Katou, K. Soda and M. Sugiyama, SAE Technical Paper Series, 2002-01-1724 Search PubMed.
- N. Hicky, P. Fornasiero, J. Kašper, M. Graziani, G. Martra, S. Coluccia, S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 209, 271 CrossRef
- K. Shimizu, T. Higashimata, M. Tsuzuki and A. Satsuma, J. Catal., 2006, 239, 117 CrossRef CAS
- K. Arve, F. Klinstedt, K. Eranen, L.-E. Lindfors and D. Yu. Murzin, Catal. Lett., 2005, 105, 133 CrossRef CAS
- K. Eränen, L.-E . Linffors, K. Klingstedt and D. Y. Murzin, J. Catal., 2003, 219, 25 CrossRef CAS
- J. Shibata, K. Shimizu, S. Satokawa, A. Satsuma and T. Hattori, Phys. Chem. Chem. Phys., 2003, 5, 2154 RSC
- J. Shibata, Y. Takada, A. Shichi, S. Satokawa, A. Satsuma and T. Hattori, J. Catal., 2004, 222, 368 CrossRef CAS
- J. Shibata, K. Shimizu, Y. Takada, A. Shichi, H. Yoshida, S. Satokawa, A. Satsuma and T. Hattori, J. Catal., 2004, 227, 367 CrossRef CAS
- J. Shibata, Y. Takada, A. Shichi, S. Satokawa, A. Satsuma and T. Hattori, Appl. Catal., B, 2004, 54, 137 CrossRef CAS
- A. Satsuma, J. Shibata, K. Shimizu and T. Hattori, Catal. Surv. Asia, 2005, 9, 75 CrossRef CAS
- G. B. F. Seijger, P. van Kooten Niekerk, K. Krishna, H. P. A. Calis, H. van Bekkum and C. M. van den Bleek, Appl. Catal., B, 2002, 40, 31
- K. Shimizu, M. Hashimoto, J. Shibata, T. Hattori and A. Satsuma, Catal. Today Search PubMed
- S. Satokawa, Chem. Lett., 2000, 294 CAS
- S. Satokawa, J. Shibata, K. Shimizu, A. Satsuma and T. Hattori, Appl. Catal., B, 2003, 42, 179 CrossRef CAS
- H. Backman, J. Jensén, F. Klinstedt, T. Salmi and D. Yu. Murzin, Appl. Catal., A, 2005, 294, 49 CrossRef CAS
- R. Burch, J. P. Breen, C. J. Hill, B. Krutzsch, B. Konrad, E. Jobson, L. Cider, K. Eranen, F. Klingstedt and L.-E. Lindfors, Top. Catal., 2004, 30, 19 CrossRef
- J. P. Breen, R. Burch, C. J. Hill, F. C. Meunier, B. Krutzsch, B. Konrad, E. Jobson, L. Cider, K. Eränen, F. Klingstedt and L.-E. Lindfors, Abst. 13th Int. Congr. Catal.,, 2004, P6-094 Search PubMed
- P. Sazama, L. Čapek, H. Drobná, Z. Sobalík, J. Dědeček, K. Arve and B. Wichterlová, J. Catal., 2005, 232, 302 CrossRef CAS
- B. Wichterlová, P. Sazama, L. Čapek, J. Dědeček, H. Drobná, Z. Sobalík and D. Kauckeý, Abst. 13th Int. Congr. Catal., 2004 Search PubMed
- K. Arve, F. Klingstedt, K. Eränen, D. Yu. Murzin, E. A. Popov and J. Eloranta, Abst. 13th Int. Congr. Catal., 2005 Search PubMed
- M. Richter, U. Bentrup, R. Eckelt, M. Schneider, M.-M. Pohl and R. Fricke, Appl. Catal., B, 2004, 51, 261 CrossRef CAS
- M. Richter, R. Fricke and R. Eckelt, Catal. Lett., 2004, 94, 115 CrossRef CAS
- R. Fricke, M. Richter, U. Bentrup, A. Brückner and R. Eckelt, Abst. 13th Int. Congr. Catal., 2004, P6-103 Search PubMed
- M. Richter, R. Fricke and R. Ecklt, Top. Catal., 2004, 94, 115 CAS
- A. Satsuma, J. Shibata, A. Wada, Y. Shinozaki and T. Hattori, Stud. Surf. Sci. Catal., 2003, 145, 235 CAS
- K. Arve, E. A. Popov, F. Klingstedt, K. Eränen, L.-E. Lindfors, J. Eloranta and D. Yu. Murzin, Catal. Today, 2005, 100, 229 CrossRef CAS
- P. Sazama and B. Wichterlova, Chem. Commun., 2005, 4810 RSC
- H. Hamada, Y. Kintaichi, M. Sasaki, T. Ito and M. Tabata, Appl. Catal., 1991, 70, L15 CrossRef CAS
- E. Kikuchi and K. Yogo, Stud. Surf. Sci. Catal., 1994, 84, 1547 CAS
- C. Yokoyama and M. Misono, J. Catal., 1994, 150, 9 CrossRef CAS
- K. Shimizu, H. Kawabata, A. Satsuma and T. Hattori, Appl. Catal., B, 1998, 19, L87 CrossRef CAS
- K. Shimizu, H. Kawabata, A. Satsuma and T. Hattori, J. Phys. Chem. B, 1999, 103, 5240 CrossRef CAS
- K. Shimizu, H. Kawabata, H. Maeshima, A. Satsuma and T. Hattori, J. Phys. Chem. B, 2000, 104, 2885 CrossRef CAS
- A. Satsuma and K. Shimizu, Prog. Energy Combust. Sci., 2003, 29, 71 CrossRef CAS
- T. Furusawa, L. Lefferts, K. Seshan and K. Aika, Appl. Catal., B, 2003, 42, 25 CrossRef CAS
- A. Iglesias-Juez, A. B. Hungría, A. Martínez-Arias, A. Fuerte, M. Fernández-Garcia, J. A. Anderson, J. C. Conesa and J. Soria, J. Catal., 2003, 217, 310 CAS
- C. Shi, M. Cheng, Z. Qu, X. Yang and X. Bao, Appl. Catal., B, 2002, 36, 173 CrossRef CAS
- C. Shi, M. Cheng, Z. Qu and X. Bao, Appl. Catal., B, 2004, 51, 171 CrossRef CAS
- Y. H. Yeom, M. Li, W. M. H. Sachtler and E. Weitz, J. Catal., 2006, 238, 100 CrossRef CAS
- Y. Yu, H. He, Q. Feng, H. Gao and X. Yang, Appl. Catal., B, 2004, 49, 159 CrossRef CAS
- S. Kameoka, T. Chafik, Y. Ukisu and T. Miyadera, Catal. Lett., 1988, 55, 211
- N. Bion, J. Sausseym, M. Haneda and M. Daturi, J. Catal., 2003, 217, 47 CAS
- Y. Li and J. Villani, Abst. 13th Int. Congr. Catal., 2005 Search PubMed
- F. Klingstedt, K. Eränen, L.-E. Lindfors, S. Andersson, L. Cider, C. Landberg, E. Jobson, L. Eriksson, T. Ilkenhans and D. Webster, Top. Catal., 2004, 30/31, 27 CrossRef
- T. Furusawa, K. Seshan, J. A. Lercher, L. Lefferts and K. Aika, Appl. Catal., B, 2002, 37, 205 CrossRef CAS
- M. C. Kung and H. H. Kung, Top. Catal., 2000, 10, 21 CrossRef CAS
- X. She and M. Flyzani-Stephanopoulos, J. Catal., 2006, 237, 79 CrossRef CAS
- N. Bogdanchikova, F. C. Meunier, A. Avalos-Borja, J. P. Breen and A. Pestryakov, Appl. Catal., B, 2002, 36, 287 CrossRef CAS
- A. Iglesias-Juez, M. Fernández-Garcia, A. Martínez-Arias, A. Schay, Zs. Koppány, A. B. Hungría, A. Fuerte, J. A. Anderson, J. C. Conesa and J. Soria, Top. Catal., 2004, 30/31, 65 CrossRef
- K. Sato, T. Yoshinari, Y. Kintaichi, M. Haneda and H. Hamada, Catal. Commun., 2003, 4, 315 CrossRef CAS
- J. P. Breen, R. Burch, C. Hardacre and C. J. Hill, J. Phys. Chem. B, 2005, 109, 4805 CrossRef CAS
- N. Aoyama, K. Yoshida, A. Abe and T. Miyadera, Catal. Lett., 1997, 43, 249 CrossRef CAS
- H. Beyer, P. A. Jacobs and J. B. Uytterhoeven, J. Chem. Soc., Faraday Trans. 1, 1976, 72, 674 RSC
- M. D. Baker, G. A. Ozin and J. Godber, J. Phys. Chem., 1985, 89, 305 CrossRef CAS
- T. Baba, N. Akinaka, M. Nomura and Y. Ono, J. Chem. Soc., Faraday Trans., 1993, 89, 595 RSC
- Z. Qu, W. Huang, M. Cgheng and X. Bao, J. Phys. Chem. B, 2005, 109, 15842 CrossRef CAS
- C. Chi, M. Cheng, Z. Qu and X. Bao, Appl. Catal., B, 2004, 51, 171 CrossRef CAS
- K. A. Bethke and H. H. Kung, J. Catal., 1997, 172, 93 CrossRef CAS
- Z. M. Wang, M. Yamaguchi, I. Goto and M. Kumagai, Phys. Chem. Chem. Phys., 2000, 2, 3007 RSC
- N. E. Bogdanchikova, M. N. Dulin, A. V. Toktarev, G. B. Shevnina, V. N. Kolomiichuk, V. I. Zaikovskii and V. P. Petranovskii, Stud. Surf. Sci. Catal., 1994, 84, 1067 CAS
- M. Matsuoka, E. Matsuda, K. Tsuji, H. Yamashita and M. Anpo, J. Mol. Catal. A: Chem., 1996, 107, 399 CrossRef CAS
- J. Texter, T. Gonsiorowski and R. Kellerman, Phys. Rev. B, 1981, 23, 4407 CrossRef CAS
- P. Mulvaney and A. Henglein, J. Phys. Chem., 1990, 94, 4182 CrossRef CAS
- T. Linnert, P. Mulvaney, A. Henglein and H. Weller, J. Am. Chem. Soc., 1990, 112, 4657 CrossRef CAS
- J. Texter, J. J. Hastreiter and J. L. Hall, J. Phys. Chem., 1983, 87, 4690 CrossRef CAS
- D. R. Brown and L. Kevan, J. Phys. Chem., 1986, 90, 1129 CrossRef CAS
- E. Janata, J. Phys. Chem. B, 2003, 107, 7334 CrossRef CAS
- F. C. Meunier, J. P. Breen, V. Zuzaniuk, M. Olsson and J. R. H. Ross, J. Catal., 1999, 187, 493 CrossRef CAS
- N. Katada, H. Igi, J.-H. Kim and M. Niwa, J. Phys. Chem. B, 1997, 101, 5969 CrossRef CAS
- N. Katada, S. Iijima, H. Igi and M. Niwa, Stud. Surf. Sci. Catal., 1997, 105, 1227
- N. Katada, Y. Kageyama and M. Niwa, J. Phys. Chem. B, 2000, 104, 7561 CrossRef CAS
- B. Wichterlová, P. Sazama, I. P. Breen, C. J. Hill, L. Čapek and Z. Sobalík, J. Catal., 2005, 235, 195 CrossRef CAS
- E. V. Kondratenko, V. A. Kondratenko, M. Richter and R. Fricke, J. Catal., 2006, 239, 23 CrossRef CAS
- Y. Wang and K. Otsuka, J. Catal., 1995, 155, 256 CrossRef CAS
- Y. Wang, K. Otsuka and K. Ebitani, Catal. Lett., 1995, 35, 259 CrossRef CAS
- J.-S. Min, H. Ishige, M. Misono and N. Mizuno, J. Catal., 2001, 198, 116 CrossRef CAS
- Y. Wang and K. Otsuka, J. Catal., 1997, 171, 106 CrossRef CAS
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566 CrossRef CAS
- W. Laufer and W. F. Hoelderlich, Chem. Commun., 2002, 1684 RSC
- M. Richter, M. Langpape, S. Kolf, G. Grubert, R. Eckelt, J. Radnik, M. Schneider, M.-M. Pohl and R. Fricke, Appl. Catal., B, 2002, 36, 261 CrossRef CAS
- M. D. Baker, G. A. Ozin and J. Godber, J. Phys. Chem., 1985, 89, 305 CrossRef CAS
- Y. Kim and K. Seff, J. Phys. Chem., 1987, 91, 668 CrossRef CAS
- T. Baba, N. Komatsu, H. Sawada, Y. Yamaguchi, T. Takahashi, H. Sugisawa and Y. Ono, Langmuir, 1999, 15, 7894 CrossRef CAS
| This journal is © the Owner Societies 2006 |