Supramolecular aggregation in diimine adducts of zinc(II) dithiophosphates: controlling the formation of monomeric, dimeric, polymeric (zig-zag and helical), and 2-D motifs
Danlin
Chen
a,
Chian Sing
Lai
a and
Edward R. T.
Tiekink
*b
aDepartment of Chemistry, National University of Singapore, Singapore 117543
bDepartment of Chemistry, The University of Texas at San Antonio, San Antonio, Texas 78248-0698, USA. E-mail: Edward.Tiekink@utsa.edu; Fax: +1 210 458 7428; Tel: +1 210 458 5774
First published on 6th December 2005
Abstract
The interplay between steric demands of dithiophosphate-bound R groups in Zn(S2P(OR)2)2 on the one hand and the coordination requirements of a variety of di-pyridyl-type bases on the other, is shown to be pivotal in determining supramolecular aggregation patterns in a series of their adducts. Thus, the combination of sterically demanding cyclohexyl groups and the congested trans-1,2-bis(2-pyridyl)ethylene ligand leads to a mononuclear species, but replacing the cyclohexyl by the less bulky isopropyl group allows for dimer formation in the structure of [Zn(S2P(OiPr)2)2(2-NC5H4C(H)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(H)C5H4N-2)0.5]2; a similar motif is found when pyrazine is the bridging ligand. These molecules aggregate in the solid-state to form chains via C–H⋯O interactions. Zig-zag polymers are found when the somewhat less congested 2,2′-dithiopyridine and bis(4-pyridyl)amine ligands are used as the bridges; the hydrogen-bonding functionality in the latter leads to a 2-D motif. Finally, exploiting the curved nature of the 1,3-bis(4-pyridyl)propane ligand leads to the formation of helical chains.
C(H)C5H4N-2)0.5]2; a similar motif is found when pyrazine is the bridging ligand. These molecules aggregate in the solid-state to form chains via C–H⋯O interactions. Zig-zag polymers are found when the somewhat less congested 2,2′-dithiopyridine and bis(4-pyridyl)amine ligands are used as the bridges; the hydrogen-bonding functionality in the latter leads to a 2-D motif. Finally, exploiting the curved nature of the 1,3-bis(4-pyridyl)propane ligand leads to the formation of helical chains.
Introduction
Interest in 1-D, 2-D and 3-D zinc coordination complexes containing pyridine residues arises from their potential use as luminescent materials.1 This application notwithstanding, the emphasis of our studies in this field is prompted by the desire to rationalise the appearance of 1D, e.g. mono- or di-nuclear species, or 2-D, e.g. straight or zig-zag polymers.2 Previous work has demonstrated that in some cases solubility characteristics dictate the formation of polymers rather than dimers,2c so that polymeric species precipitate regardless of the ratio of zinc dithiophosphate (Zn(S2P(OR)2)2; Lewis acid) and dipyridyl-type ligand (Lewis base) employed, i.e. 1 ∶ 1 or 2 ∶ 1 in the reaction mixture. Of more interest is the ability to control polymer formation by systematically varying the nature of the dithiophosphate-bound R group so that in cases where R is small, e.g. isopropyl (iPr), polymers are found, but under the same conditions dimeric species are only formed when the steric bulk of R is increased to cyclohexyl (Cy).2 These steric-based arguments have been utilised to great effect in rationalising the appearance of different supramolecular motifs in the binary 1,1-dithiolates of the zinc-triad elements.3 Allied studies on the related cadmium dithiophosphate adducts led to similar conclusions to those noted for the zinc dithiophosphates but the ability to control polymer topology was less evident.4 For example it is possible to isolate polymeric species with 4,4′-bipyridine, i.e. [M(S2P(OR)2)2(4-NC5H4C5H4N-4)0.5]∞ for M = Cd and R = iPr and Cy,4 but when M = Zn, polymer formation was only possible when R = iPr.5a While a little disappointing, the reduced control in the cadmium-containing systems can be related to the longer cadmium-ligand bonds that mitigates steric influences. Given that greater control over polymer formation, and when formed, topology, was possible in the case of the zinc dithiophosphate system,2 it was thought of interest to explore the structural chemistry of adducts containing a wider range of di-pyridyl-type ligands, see Chart 1, with the view of elaborating upon the principles outlined above and generating novel polymer topologies for this class of compound.2,5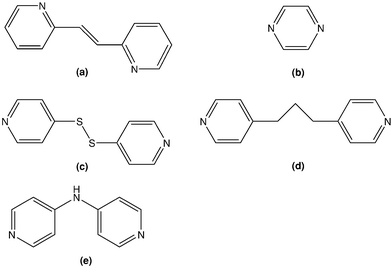 | ||
| Chart 1 Chemical structures of the di-pyridyl-type molecules used in the present study: (a) trans-1,2-bis(2-pyridyl)ethylene, (b) pyrazine, (c) 2,2′-dithiopyridine, (d) 1,3-bis(4-pyridyl)propane, and (e) bis(4-pyridyl)amine. | ||
Results and discussion
Monomeric species
The only mononuclear species described herein is found in the structure of [Zn(S2P(OCy)2)2(2-NC5H4C(H)C(H)C5H4N-2)]
(1), as illustrated in Fig. 1; key geometric parameters for this and all other structures described are collected in Table 1.
![Coordination geometry for the zinc centre in the mononuclear structure of [Zn(S2P(OCy)2)2(2-NC5H4C(H)C(H)C5H4N-2)] (1).](/image/article/2006/CE/b513393a/b513393a-f1.gif) | ||
| Fig. 1 Coordination geometry for the zinc centre in the mononuclear structure of [Zn(S2P(OCy)2)2(2-NC5H4C(H)C(H)C5H4N-2)] (1). | ||
C(H)C5H4N-2)]
(1), [Zn(S2P(OiPr)2)2(2-NC5H4C(H)C(H)C5H4N-2)0.5]2
(2), [Zn(S2P(OiPr)2)2(1,4-NC4H4N)0.5]2
(3), [Zn(S2P(OiPr)2)2(4-NC5H4SSC5H4N-4)]∞
(4), [Zn(S2P(OiPr)2)2(4-NC5H4(CH2)3C5H4N-4)]∞
(5), and [Zn(S2P(OiPr)2)2(4-NC5H4N(H)C5H4N-4)]∞
(6)
| 5 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | 1 | 2 | 3 | 4 | Molecule ab | Molecule bb | 6 |
| a Where applicable, see figure captions for symmetry operations. b Two independent molecules comprise the asymmetric unit. For the first molecule, a, N1 is N2, N2 is N4, and for the second, b, add “4” to each S label, “2” to each P label, and N2 is N3. | |||||||
| Zn–S1 | 2.3652(19) | 2.3162(7) | 2.3320(12) | 2.284(3) | 2.3092(12) | 2.3101(13) | 2.3039(12) |
| Zn–S2 | 2.4513(19) | 2.6862(8) | 2.4361(13) | 3.912(3) | 3.8066(13) | 3.7262(14) | 4.0715(14) |
| Zn–S3 | 2.265(2) | 2.3213(7) | 2.2563(11) | 2.316(2) | 2.3052(15) | 2.3199(11) | 2.2950(14) |
| Zn–S4 | 3.373(2) | 2.8740(8) | 3.2463(15) | 5.211(3) | 3.7854(16) | 3.8579(15) | 3.5288(16) |
| Zn–N1 | 2.084(6) | 2.0494(19) | 2.072(3) | 2.080(6) | 2.022(3) | 2.057(3) | 2.054(4) |
| Zn–N2a | 4.977(8) | — | — | 2.051(6) | 2.061(3) | 2.023(3) | 2.085(4) |
| P1–S1 | 2.005(2) | 2.0140(9) | 1.9921(15) | 2.011(4) | 2.0136(15) | 2.0257(16) | 2.0195(17) |
| P1–S2 | 1.992(2) | 1.9717(9) | 1.9897(14) | 1.904(4) | 1.9420(14) | 1.9307(19) | 1.9396(17) |
| P2–S3 | 2.016(2) | 2.0165(9) | 2.0286(15) | 2.011(3) | 2.000(2) | 2.0200(15) | 2.0305(17) |
| P2–S4 | 1.944(3) | 1.9605(9) | 1.9444(16) | 1.941(3) | 1.905(3) | 1.9354(16) | 1.9372(19) |
| S1–Zn–S2 | 86.01(6) | 82.09(2) | 86.29(4) | 60.39(8) | 62.33(3) | 63.49(4) | 56.80(4) |
| S3–Zn–S4 | 70.50(6) | 77.81(2) | 73.22(4) | 28.03(6) | 61.28(5) | 60.48(3) | 66.85(4) |
| S1–Zn–S3 | 125.64(8) | 128.84(3) | 132.36(5) | 113.66(10) | 97.33(5) | 100.09(4) | 118.76(5) |
| S2–Zn–S4 | 153.88(7) | 176.57(2) | 175.65(4) | 149.59(6) | 135.03(4) | 135.64(3) | 110.90(3) |
| N1–Zn–N2a | 82.1(2) | — | — | 99.0(2) | 103.07(13) | 106.87(12) | 96.09(15) |
The zinc atom in (1) is chelated by one dithiophosphate ligand and the second dithiophosphate coordinates effectively in the monodentate mode as the Zn⋯S4 separation is 3.373(2) Å. The disparity in the Zn–S interactions formed by these ligands is systematically reflected in both the difference in the associated P–S bond distances and the variation in the chelate angles (Table 1); this observation is mirrored in all of the structures described herein and is not discussed further. The coordination geometry about the zinc centre is completed by a nitrogen atom derived from a monodentate trans-1,2-bis(2-pyridyl)ethylene ligand; the N2 atom is separated by 4.977(8) Å from the zinc centre. The conformation of the trans-1,2-bis(2-pyridyl)ethylene ligand is one in which the pyridine-nitrogen atoms are syn as confirmed by the refinement and an examination of the bond distances within the pyridyl rings. The closest intermolecular contact of significance in the crystal structure of (1) is of the type C–H⋯O [C26–H⋯O4i is 2.59 Å, C26⋯O4i is 3.520(11) Å with an angle at H of 173° for symmetry operation i: x, 1 + y, z] that extends translationally along the b-axis to form a chain.
The structure of (1) appears to be the first crystallographically proven example in which the trans-1,2-bis(2-pyridyl)ethylene ligand coordinates in the monodentate mode.6 In the only other structure containing the trans-1,2-bis(2-pyridyl)ethylene ligand in which only one of the pyridine-nitrogen atoms coordinates a metal centre, i.e.
[(CO)4Re(2-NC5H4C(H)C(H)C5H4N-2)Re(CO)4], the central double bond coordinates the second rhenium atom.7 This coordination mode also features a syn arrangement of the bridging ligand and a syn, bidentate bridging conformation is found in the structure of [(CO)4Re(2-NC5H4C(H)C(H)C5H4N-2)Re(CO)4]·C6H12, but again the double bond coordinates a rhenium atom.7 Otherwise, the overwhelming majority of coordination complexes containing the trans-1,2-bis(2-pyridyl)ethylene ligand feature an anti conformation.6 In (1), the NS3 donor set defines a distorted tetrahedral geometry. In this description, the range of angles subtended at the zinc atom is 86.01(6) to 125.64(8)° with the wider S1–Zn–S3 angle clearly due to the close approach of the S4 atom. If the S4 atom was considered to be bonding, the geometry would be best described as distorted trigonal bipyramidal with the axial positions being occupied by the S2 and S4 atoms. In the context of the present study, the key point is that only a monomeric species could be generated from solutions containing a 1 ∶ 1 ratio of zinc dithiophosphate and trans-1,2-bis(2-pyridyl)ethylene when, based on stoichiometry, a polymer might have been anticipated. However, it proved possible to generate a dimeric compound of trans-1,2-bis(2-pyridyl)ethylene when the steric bulk of R was reduced, i.e. when R = iPr.
The dimeric structure of [Zn(S2P(OiPr)2)2(2-NC5H4C(H)C(H)C5H4N-2)0.5]2
(2), Fig. 2 and Table 1, was obtained from solutions containing ratios of Zn(S2P(OiPr)2)2 to ligand of 1 ∶ 1 and 2 ∶ 1 (based on comparative infrared data) and indicates that again a polymeric species could not be obtained. The molecule is centrosymmetric, implying an anti disposition of the 2-pyridyl groups, and features a coordination geometry similar to that just described for (1) but as the Zn–S4 distance of 2.8740(8)
Å is 0.5 Å shorter and the S2–Zn–S4 angle closer to 180° compared to the situation in (1), a trigonal bipyramidal geometry is a better description for the zinc atom geometry. A chain motif is also formed in the crystal structure of (2), mediated in the first instance by C–H⋯O interactions. Thus, C16–H⋯O4ii is 2.54 Å, C16⋯O4ii is 3.449(3)
Å with an angle of 163° at H for symmetry operation ii: 1 +
x, y, z. The chain is aligned along the a-axis and associates into a double chain via
π⋯π interactions; the distance of the ring centroid of (N1, C13–C17) to its centrosymemtric counterpart is 3.72 Å
(symmetry operation 1 −
x, −y, 1 −
z).
![Coordination geometry for the zinc centre in the dinuclear structure of [Zn(S2P(OiPr)2)2(2-NC5H4C(H)C(H)C5H4N-2)0.5]2 (2). Click here to access a 3D image of Fig. 2.](/image/article/2006/CE/b513393a/b513393a-f2.gif) | ||
| Fig. 2 Coordination geometry for the zinc centre in the dinuclear structure of [Zn(S2P(OiPr)2)2(2-NC5H4C(H)C(H)C5H4N-2)0.5]2 (2). Click /ej/ce/2006/b513393a/2.htm to access a 3D image of Fig. 2. | ||
The final monomeric structure to be described is that of [Zn(S2P(OiPr)2)2(1,4-NC4H4N)0.5]2 (3), Fig. 3 and Table 1, containing the small molecule, pyrazine, as the bridging ligand. The molecule is centrosymmetric and adopts a coordination geometry closely resembling that described in (1) above. As with (2), it was not possible to isolate a polymer from solutions containing a 1 ∶ 1 ratio of Zn(S2P(OiPr)2)2 and pyrazine (based on infrared data). A chain motif is discernable in the crystal structure of (3), the weak nature of the C–H⋯O interactions mediating this chain notwithstanding. Centrosymmetrically related dimers associate via C5–H⋯O3ii interactions of 2.75 Å so that C5⋯O3ii is 3.527(9) Å and the angle at H is 154° for symmetry operation ii: −x, −y, 1 − z. The chains thus formed are aligned along the c-axis.
![Coordination geometry for the zinc centre in the dinuclear structure of [Zn(S2P(OiPr)2)2(1,4-NC4H4N)0.5]2 (3).](/image/article/2006/CE/b513393a/b513393a-f3.gif) | ||
| Fig. 3 Coordination geometry for the zinc centre in the dinuclear structure of [Zn(S2P(OiPr)2)2(1,4-NC4H4N)0.5]2 (3). | ||
The pair of structures, (1) and (2), allows two conclusions to be made. Firstly, the preclusion of polymeric structure is not due to cyclohexyl versus isopropyl but most likely arises owing to the inability of the trans-1,2-bis(2-pyridyl)ethylene ligand to bridge successive Zn(S2P(OR)2)2 entities owing to the short Zn–N (and Zn–S) interactions that would bring the various components of the molecules too close together. When these constraints do not exist, as in the cadmium analogues, where for example the Cd–N separations are greater than 2.5 Å, both the R = iPr and Cy compounds form zig-zag polymers.4 Secondly, and suporting the above argument, a putative dimeric structure similar to (2) but with R = Cy is precluded owing to steric reasons associated with the dithiophosphate-bound R groups. Rotating molecule (2) and substituting R = iPr for R = Cy, indicates that impossible steric clashes would arise in the latter precluding its formation, even in this case where the trans-1,2-bis(2-pyridyl)ethylene ligands adopt an anti conformation.
The underlying assumption of this systematic study is that each reported crystal structure is truly representative of the bulk isolated material. While both spectroscopic and microanalytical evidence supports this assumption, see Experimental section, the possibility of other structures with similar crystal habits can not be absolutely discounted.
Polymeric species
The first polymeric structure to be described is that containing the 2,2′-dithiopyridine ligand, [Zn(S2P(OiPr)2)2(4-NC5H4SSC5H4N-4)]∞ (4); Fig. 4 and Table 1. The dithiophosphate ligands are effectively monodentate and as the di-pyridyl-type ligand is bridging, the coordination geometry is based on a tetrahedron defined by a N2S2 donor set. The tetrahedral angles span a relatively narrow range of 99.1(2)°, for N1–Zn–N2i, to 119.68(19)° for N1 Zn S1, consistent with the rather long Zn⋯S2 and Zn⋯S4 secondary interactions of 3.914(3) and 5.206(3) Å, respectively. The conformational flexibility associated with the 2,2′-dithiopyridine ligand in terms of the ready availability of two nitrogen atoms for coordination, as well as the kink in the molecule owing to the disulfide bridge, allows for the formation of a polymeric structure with a zig-zag topology as shown in the two views in Fig. 5.![Coordination geometry for the zinc centre in the polymeric structure of [Zn(S2P(OiPr)2)2(4-NC5H4SSC5H4N-4)]∞ (4). Symmetry operation i: x, 1 −
y, ½
+
z.](/image/article/2006/CE/b513393a/b513393a-f4.gif) | ||
| Fig. 4 Coordination geometry for the zinc centre in the polymeric structure of [Zn(S2P(OiPr)2)2(4-NC5H4SSC5H4N-4)]∞ (4). Symmetry operation i: x, 1 − y, ½ + z. | ||
![Two views of the polymeric structure of [Zn(S2P(OiPr)2)2(4-NC5H4SSC5H4N-4)]∞ (4). (a) Viewed down the a-direction and (b) viewed approximately down the c-direction. Click here to access a 3D image of Fig. 5b.](/image/article/2006/CE/b513393a/b513393a-f5.gif) | ||
| Fig. 5 Two views of the polymeric structure of [Zn(S2P(OiPr)2)2(4-NC5H4SSC5H4N-4)]∞ (4). (a) Viewed down the a-direction and (b) viewed approximately down the c-direction. Click /ej/ce/2006/b513393a/5b.htm to access a 3D image of Fig. 5b. | ||
The polymer is propagated by glide symmetry along the c-direction. Zig-zag topologies are the common polymeric form for zinc dithiophosphates and so in that sense, the observed structure is entirely consistent with expectation.2,5 The Zn⋯Zn and N1⋯N2 distances are 11.04 and 7.80 Å, respectively and the pyridyl rings are effectively orthogonal as seen in the dihedral angle of 89.3(4)° between their respective least-squares planes. When the nature of the bridging di-pyridyl ligand is changed to the somewhat curved4,6 1,2-bis(4-pyridyl)propane ligand, a new polymer topology, unprecedented for the zinc (and cadmium) dithiophosphates, is found.
Two independent but virtually identical formula units comprise the crystallographic asymmetric unit of [Zn(S2P(OiPr)2)2(4-NC5H4(CH2)3C5H4N-4)]∞ (5). The immediate coordination geometry for each zinc atom is shown in Fig. 6 and selected geometric parameters are collated in Table 1. The zinc atom geometries resemble closely that described for (4) above, with the ranges of tetrahedral angles being 97.33(5) to 118.85(12)° and 100.09(4) to 117.26(10)°, respectively for the two independent zinc atom geometries. The polymer is propagated along the a-axis by translational symmetry but nevertheless adopts a helical topology, see Fig. 7. This arises as the somewhat curved 1,2-bis(4-pyridyl)propane links effectively wrap around the [Zn(S2P(OiPr)2)2]∞ backbone in a concerted fashion. A full turn of the helix corresponds to two independent formula units and the unit cell edge a, i.e. 18.5675(6) Å. The flexibility of the 1,3-bis(4-pyridyl)propane ligand to span the zinc atoms is evidenced by the Zn⋯Zn separations of 13.20 and 12.25 Å, for molecules a and b, respectively, i.e. that differ by nearly 1 Å, and N⋯N separations of 9.67 and 9.00 Å, respectively. The dihedral angles between the two pyridyl planes for the independent ligands are 64.1(2) and 72.4(2)°, respectively.
![Coordination geometry for the two independent zinc centres in the polymeric structure of [Zn(S2P(OiPr)2)2(4-NC5H4(CH2)3C5H4N-4)]∞ (5). Symmetry operation i: 1 +
x, y, z and ii: −1 +
x, y, z.](/image/article/2006/CE/b513393a/b513393a-f6.gif) | ||
| Fig. 6 Coordination geometry for the two independent zinc centres in the polymeric structure of [Zn(S2P(OiPr)2)2(4-NC5H4(CH2)3C5H4N-4)]∞ (5). Symmetry operation i: 1 + x, y, z and ii: −1 + x, y, z. | ||
![Two views of the polymeric structure of [Zn(S2P(OiPr)2)2(4-NC5H4(CH2)3C5H4N-4)]∞ (5). (a) Viewed down the c-direction and (b) viewed approximately down the a-direction. Click here to access a 3D image of Fig. 7.](/image/article/2006/CE/b513393a/b513393a-f7.gif) | ||
| Fig. 7 Two views of the polymeric structure of [Zn(S2P(OiPr)2)2(4-NC5H4(CH2)3C5H4N-4)]∞ (5). (a) Viewed down the c-direction and (b) viewed approximately down the a-direction. Click /ej/ce/2006/b513393a/7.htm to access a 3D image of Fig. 7. | ||
The final structure to be described is that of [Zn(S2P(OiPr)2)2(4-NC5H4N(H)C5H4N-4)]∞ (6), i.e. with the bis(4-pyridyl)amine ligand. The coordination geometry in (6), Fig. 8 and Table 1, is in essential agreement with those found in (4) and (5); the range of tetrahehedral angles is 96.09(15) to 118.76(5)°. A zig-zag polymer topology is found that is propagated by glide symmetry along the c-axis. The Zn⋯Zn and N⋯N separations are 11.69 and 7.77 Å, respectively and the rather flattened nature of the dipyridyl ligand is emphasised in the dihedral angle between the two rings of only 33.9(2)°, compared to above. To a first approximation, the polymeric structure of (6) is the same as that found for (4) and might be considered to represent another example of a zig-zag polymer for this class of compound. While this is true, interest in the structure of (6) stems from the ability of the bis(4-pyridyl)amine ligand to engage in further supramolecular association by virtue of its acidic amine-hydrogen atom. As shown in Fig. 9, the N3-H atom does indeed form a hydrogen-bonding interaction with a symmetry related O1 atom so that H⋯O1ii is 2.46 Å, N3⋯O1ii is 3.300(5) Å and the angle subtended at H is 164° for symmetry operation ii: x, −y, ½ + z. From the imposed glide symmetry, these interactions occur on both sides of the polymer which results in the formation of a 2-D array that stacks along the a-direction, see Fig. 9.
![Coordination geometry for the zinc centre in the polymeric structure of [Zn(S2P(OiPr)2)2(4-NC5H4N(H)C5H4N-4)]∞ (6). Symmetry operation i: x, 1 −
y, −½
+
z.](/image/article/2006/CE/b513393a/b513393a-f8.gif) | ||
| Fig. 8 Coordination geometry for the zinc centre in the polymeric structure of [Zn(S2P(OiPr)2)2(4-NC5H4N(H)C5H4N-4)]∞ (6). Symmetry operation i: x, 1 − y, −½ + z. | ||
![Three views of the polymeric structure of [Zn(S2P(OiPr)2)2(4-NC5H4N(H)C5H4N-4)]∞ (6). (a) Viewed down the a-direction and (b) hydrogen-bonded 2-D sheets viewed down the a-direction, and (c) 2-D layers viewed approximately down the b-direction. Click here to access a 3D image of Fig. 9.](/image/article/2006/CE/b513393a/b513393a-f9.gif) | ||
| Fig. 9 Three views of the polymeric structure of [Zn(S2P(OiPr)2)2(4-NC5H4N(H)C5H4N-4)]∞ (6). (a) Viewed down the a-direction and (b) hydrogen-bonded 2-D sheets viewed down the a-direction, and (c) 2-D layers viewed approximately down the b-direction. Click /ej/ce/2006/b513393a/9.htm to access a 3D image of Fig. 9. | ||
Conclusions
Steric factors associated with the dithiophospate-bound R groups and/or various di-pyridyl-type ligands can be exploited so as to preclude dimer and polymer formation, as exemplified in the structures of (1)–(3). Reducing the steric constraints of the di-pyridyl-type ligands allows for polymer formation and judicious choice allows control of the resultant polymer topology, i.e. zig-zag for (4) and (6) versus helical for (5). The addition of further supramolecular capability, such as hydrogen-bonding potential, allows for the formation of more sophisticated aggregates such as 2-D sheets in the case of (6). A clear advantage of the controlling aggregate size and/or topology of the zinc dithiophosphates adducts with di-pyridyl-type ligands is the reduced distances of the zinc to ligand interactions that make these species more susceptible to steric control. Thus, similar steric control in the analogous cadmium dithiophosphates is not nearly as great.4 While the sensitivity of zinc dithiophosphate di-pyridyl-type ligand adducts to such steric control is gratifying, it also indicates that the general applicability of such principles to other systems may be limited to those that form similarly short metal–ligand bonds that would accenuate the “steric effect for supramolecular aggregation”.Experimental
Synthesis and characterisation
The Zn(S2P(OR)2)2, R = iPr and Cy, compounds were prepared in high yields from the reaction of Zn(NO3)2·6H2O (Merck) and the respective ammonium dithiophosphate (Cheminova) in aqueous solution. The adducts were obtained from refluxing (2 h) the parent zinc compound (0.2 g) with either a 1 or 2 stoichiometric amount of trans-1,2-bis(2-pyridyl)ethylene, pyrazine, 2,2′-dithiopyridine, 1,3-bis(4-pyridyl)propane (Aldrich) and bis(4-pyridyl)amine in CHCl3 solution; the ligand bis(4-pyridyl)amine, a known compound,8 was prepared simply by reacting 4-pyridylamine (Aldrich) with 4-pyridinecarboxaldehye (Aldrich) in ethanol solution under reflux. After reaction, the solvent was removed in vacuo and the residue recrystallised by slow evaporation from dichloromethane–hexane (2 : 1) for (1) and (3), chloroform–acetonitrile (3 : 1) for (2), (4) and (5), and chloroform–methanol (1 : 1) for (6) solutions of the respective compounds. Infrared data were recorded as KBr discs on a Excalibur Series Bio-Rad Merlin FTS 3000 spectrophotometer. Microanalytical data were measured on a Perkin-Elmer PE 2400 CHN Elemental Analyzer for dried samples.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C(H)C5H4N-2)]
(1).
Obs. (Calc.): C, 51.61 (51.82); H, 6.42 (6.52%). IR (KBr, cm−1): 1152 (m)
ν(C–O); 976 (s)
ν(P–O); 675 (m)
νasym(P–S); 577 (m)
νsym(P–S); mp 145–146 °C.
C(H)C5H4N-2)0.5]2
(2).
Obs. (Calc.): C, 37.14 (37.08); H, 5.75 (5.71%). IR (KBr, cm−1): 1105 (m)
ν(C–O); 975 (s)
ν(P–O); 664 (s)
νasym(P–S); 545 (m)
νsym(P–S); mp 133–134 °C.
C(H)C5H4N-2)]
(1).
Obs. (Calc.): C, 51.61 (51.82); H, 6.42 (6.52%). IR (KBr, cm−1): 1152 (m)
ν(C–O); 976 (s)
ν(P–O); 675 (m)
νasym(P–S); 577 (m)
νsym(P–S); mp 145–146 °C.
C(H)C5H4N-2)0.5]2
(2).
Obs. (Calc.): C, 37.14 (37.08); H, 5.75 (5.71%). IR (KBr, cm−1): 1105 (m)
ν(C–O); 975 (s)
ν(P–O); 664 (s)
νasym(P–S); 545 (m)
νsym(P–S); mp 133–134 °C.
Crystallography
Intensity data were measured at 223(2) K on a Bruker SMART CCD diffractometer employing Mo-Kα radiation. Data processing and absorption correction were accomplished with SAINT9 and SADABS,10 respectively. The structures were solved by heavy-atom methods11 and refinement (anisotropic displacement parameters, hydrogen atoms in the riding model approximation and a weighting scheme of the form w = 1/[σ2(Fo2) + aP2] for P = (Fo2 + 2Fc2)/3) was on F2.12 The structure of (1) was refined as a racemic twin precluding the determination of the absolute structure, but that of (6) was indicated by the value of the Flack parameter, i.e. −0.009(12).13 As might be anticipated in systematic studies of this type, some of the crystals examined were not optimal resulting in difficulties in their refinements. Thus, significant thermal motion was evident in the refinement of (4) so that isopropyl-carbon atoms were refined isotropically. Those for the O2-isopropyl group were resolved over two positions with site occupancy factors of 0.77(2) ∶ 0.23(2). Similarly, in both (5) and (6) high thermal motion was noted. For (5), it was possible to resolve the C16–C18 atoms into two sites with the major component (isotropic refinement) having a site occupancy factor = 0.636(13). In the case of (6), the C9 atom was resolved into two sites with the major component (isotropic refinement) having a site occupancy factor = 0.59(2). The largest residual electron density peaks in the refinements of (1), (3) and (5) were in the vicinity of the zinc and the weakly bound S4 atom and that in (4) was located near the S5 atom. Crystallographic data and final refinement details are given in Table 2. Fig. 1–4, 6 and 8 were drawn with ORTEP14 at the 50% probability level and the remaining figures were drawn with the DIAMOND programme.15 When applicable, in all figures only the major component of the disordered atoms is shown. Data manipulation and analysis was with teXsan16 and PLATON.17CCDC reference numbers 284517–284522. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b513393a
C(H)C5H4N-2)]
(1), [Zn(S2P(OiPr)2)2(2-NC5H4C(H)C(H)C5H4N-2)0.5]2
(2), [Zn(S2P(OiPr)2)2(1,4-NC4H4N)0.5]2
(3), [Zn(S2P(OiPr)2)2(4-NC5H4SSC5H4N-4)]∞
(4), [Zn(S2P(OiPr)2)2(4-NC5H4(CH2)3C5H4N-4)]∞
(5), and [Zn(S2P(OiPr)2)2(4-NC5H4N(H)C5H4N-4)]∞
(6)
| Compound | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Formula | C36H54N2O4P2S4Zn | C36H66N2O8P4S8Zn2 | C14H30NO4P2S4Zn | C22H36N2O4P2S6Zn | C25H42N2O4P2S4Zn | C22H37N3O4P2S4Zn |
| Formula weight | 834.36 | 1166.01 | 531.94 | 712.20 | 690.16 | 663.10 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Orthorhombic | Monoclinic | Monoclinic |
| Space group | P21 | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Ccca | P21/c | Pc |
| a/Å | 9.7269(8) | 7.8735(8) | 8.3237(14) | 21.988(5) | 18.5675(6) | 12.1969(7) |
| b/Å | 8.6585(7) | 17.7046(15) | 11.2642(19) | 35.283(7) | 23.9914(7) | 8.3847(5) |
| c/Å | 24.853(2) | 20.0325(19) | 14.950(4) | 17.564(3) | 16.1405(5) | 15.5757(10) |
| α/° | 90 | 90 | 77.797(3) | 90 | 90 | 90 |
| β/° | 100.872(2) | 90.638(2) | 73.836(3) | 90 | 103.262(1) | 95.676(2) |
| γ/° | 90 | 90 | 68.317(3) | 90 | 90 | 90 |
| V/Å3 | 2055.6(3) | 2792.3(5) | 1241.8(4) | 13626(5) | 6998.2(4) | 1585.08(17) |
| Z | 2 | 2 | 2 | 16 | 8 | 2 |
| D c/g cm−3 | 1.348 | 1.387 | 1.423 | 1.389 | 1.310 | 1.389 |
| F(000) | 880 | 1216 | 554 | 5920 | 2896 | 692 |
| μ(MoKα)/mm−1 | 0.917 | 1.316 | 1.472 | 1.211 | 1.062 | 1.170 |
| 2θmax/° | 60.2 | 60.0 | 60.2 | 60.0 | 60.2 | 60.0 |
| Measured data | 17302 | 23156 | 11752 | 54255 | 59094 | 13069 |
| Unique data | 9963 | 8112 | 7146 | 9885 | 20496 | 7130 |
| Observed data (I ≥ 2.0σ(I)) | 7925 | 5566 | 4755 | 4186 | 11846 | 5186 |
| Variables | 442 | 271 | 235 | 287 | 683 | 325 |
| R, obs. data; all data | 0.078; 0.100 | 0.047; 0.079 | 0.065; 0.101 | 0.108; 0.212 | 0.074; 0.124 | 0.051; 0.065 |
| a; b in weighting scheme | 0.097; 6.644 | 0.051; 0 | 0.105; 0.636 | 0.200; 0 | 0.101; 6.119 | 0.072; 0 |
| Rw, obs. data; all data | 0.209; 0.223 | 0.100; 0.113 | 0.165; 0.191 | 0.324; 0.390 | 0.192; 0.223 | 0.118; 0.128 |
| Largest residual/e Å−3 | 1.24 | 0.46 | 2.21 | 1.41 | 1.62 | 0.90 |
| CCDC deposition no. | 284517 | 284518 | 284519 | 284520 | 284521 | 284522 |
Acknowledgements
The University of Singapore is thanked for a research grant (R-143-000-151-112) in support of this research. We also thank Cheminova, for the gift of the ammonium dithiophosphates, and Prof. J. J. Vittal, for the gift of 2,2′-dithiopyridine, used in this research programme.References
- (a) K.-Y. Ho, W.-Y. Yu, K.-K. Cheung and C.-M. Che, J. Chem. Soc., Dalton Trans., 1999, 1581 RSC; (b) N. W. Alcock, P. R. Barker, J. M. Haider, M. J. Hannon, C. L. Painting, Z. Pikramenou, E. A. Plummer, K. Rissanen and P. Saarenketo, J. Chem. Soc., Dalton Trans., 2000 Search PubMed; (c) Q. Wu, J. A. Lavigne, Y. Tao, M. D'Iorio and S. Wang, Inorg. Chem., 2000, 39, 5248 CrossRef CAS; (d) V. W.-W. Yam, Y.-L. Pui and K.-K. Cheung, Inorg. Chem., 2000, 39, 5741 CrossRef CAS; (e) J. Tao, M.-L. Tong, J.-X. Shi, X.-M. Chen and S. W. Ng, Chem. Commun., 2000, 2043 RSC; (f) V. W.-W. Yam, Y.-L. Pui, K.-K. Cheung and N. Zhu, New J. Chem., 2002, 26, 536 RSC; (g) H.-F. Zhu, W. Zhao, T. Okamura, B.-L. Fei, W.-Y. Sun and N. Ueyama, New J. Chem., 2002, 36, 1277 RSC; (h) L. S. Sapochak, F. E. Benincasa, R. S. Schofield, J. L. Baker, K. K. C. Riccio, D. Fogarty, H. Kohlmann, K. F. Ferris and P. E. Burrows, J. Am. Chem. Soc., 2002, 124, 6119 CrossRef CAS; (i) M. Ghedini, M. La Deda, I. Aiello and A. Grisolia, J. Chem. Soc., Dalton Trans., 2002, 3406 RSC; (j) L.-N. Zhu, L. Z. Zhang, W.-Z. Wang, D.-Z. Liao, P. Cheng, Z.-H. Jiang and S.-P. Yan, Inorg. Chem. Commun., 2002, 5, 1017 CrossRef CAS; (k) I. V. Sazanovich, C. Kirmaier, E. Hindin, L. Yu, D. F. Bocian, J. S. Lindsey and D. Holten, J. Am. Chem. Soc., 2004, 126, 2664 CrossRef; (l) J. Zhang, S. Gao and C.-M. Che, Eur. J. Inorg. Chem., 2004, 956 CrossRef CAS; (m) S.-L. Zheng and X.-M. Chen, Aust. J. Chem., 2004, 57, 703 CrossRef CAS; (n) B. Dutta, P. Bag, U. Floerke and K. Nag, Inorg. Chem., 2005, 44, 147 CrossRef CAS; (o) X.-S. Wang, Y.-Z. Tang, X.-F. Huang, Z.-R. Qu, C.-M. Che, P. W. H. Chan and R.-G. Xiong, Inorg. Chem., 2005, 44, 5278 CrossRef CAS; (p) D. Song, W. L. Jia, G. Wu and S. Wang, Dalton Trans., 2005, 433 RSC.
- (a) C. S. Lai, Y. X. Lian, T. C. Yap and E. R. T. Tiekink, CrystEngComm, 2002, 4, 596 RSC; (b) S. F. Soh, C. S. Lai and E. R. T. Tiekink, Acta Crystallogr., Sect. E, 2002, 58, m641 CrossRef; (c) C. S. Lai, S. Liu and E. R. T. Tiekink, CrystEngComm, 2004, 6, 221 RSC; (d) D. Chen, C. S. Lai and E. R. T. Tiekink, Acta Crystallogr., Sect. E, 2005, 61, m2052.
- E. R. T. Tiekink, CrystEngComm, 2003, 5, 101 RSC.
- C. S. Lai and E. R. T. Tiekink, CrystEngComm, 2004, 6, 593 RSC.
- (a) L. A. Glinskaya, V. G. Shchukin, R. F. Klevtsova, A. N. Mazhara and S. V. Larionov, J. Struct. Chem., 2000, 41, 772; (b) D.-L. Zhu, Y.-P. Yu, G.-C. Guo, H.-H. Zhuang, J.-S. Huang, Q. Liu, Z. Xu and X.-Z. You, Acta Crystallogr., Sect. C, 1996, 52, 1963 CrossRef.
- F. H. Allen and O. Kennard, Chem. Des. Automat. News, 1993, 8, 1 & 31 Search PubMed.
- R. A. Machado, D. Rivillo, A. J. Arce, L. D'Ornelas, Y. De Sanctis, R. Atencio, T. Gonzalez and E. Galarza, J. Organomet. Chem., 2004, 689, 2486 CrossRef CAS.
- E. Koenigs and G. Jung, J. Prakt. Chem., 1933, 137, 141 CAS.
- SAINT, Version V5.6, Bruker AXS Inc., Madison, WI, 2000 Search PubMed.
- SADABS, (a) G. M. Sheldrick, University of Göttingen, Germany, 2000; (b) R. Blessing, Acta Crystallogr., Sect. A, 1995, 51, 33 CrossRef.
- P. T. Beurskens, G. Admiraal, G. Beurskens, W. P. Bosman, S. García-Granda, J. M. M. Smits and C. Smykalla, The DIRDIF program system, Technical Report of the Crystallography Laboratory, University of Nijmegen, The Netherlands, 1992 Search PubMed.
- G. M. Sheldrick, SHELXL97, Program for the Refinement of Crystal Structures, University of Göttingen, Gemany, 1997 Search PubMed.
- H. D. Flack, Acta Crystallogr., Sect. A, 1983, 39, 876 CrossRef.
- C. K. Johnson, ORTEP II, Report ORNL-5136, Oak Ridge National Laboratory, Oak Ridge, TN, 1976 Search PubMed.
- DIAMOND, Visual Crystal Structure Information System, Version 2.1e, CRYSTAL IMPACT, Bonn, Germany, 2002 Search PubMed.
- teXsan, Structure Analysis Package, Molecualr Structure Corporation, Houston, TX, 1992 Search PubMed.
- T. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2003 Search PubMed.
| This journal is © The Royal Society of Chemistry 2006 |