
“A posteriori” modification of carbosilane dendrimers and dendrons: their activation in core and branch positions
Abstract
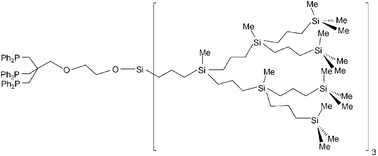
The introduction of phenyl groups at different points on carbosilane dendrimers allows their acidolytic conversion to highly reactive triflato groups which in turn are readily substituted by anionic nucleophiles. Core phenylated first–fourth generation dendrimers were synthesized from
- This article is part of the themed collection: Celebrating the 150th anniversary of the German Chemical Society
 Please wait while we load your content...
Please wait while we load your content...

