Marine isocyanides and related natural products – structure, biosynthesis and ecology
Mary J. Garsona and Jamie S. Simpsonb
aDepartment of Chemistry, The University of Queensland, Brisbane 4072 QLD, Australia
bResearch School of Chemistry, Australian National University, Canberra 0200 ACT, Australia
First published on 14th January 2004
Abstract
Covering: up to mid 2003. Previous review: Nat. Prod. Rep., 1988, 5, 229
This review highlights structural and biosynthetic work on a group of nitrogen-functionalised terpenes that are almost exclusively found in marine invertebrates and the animals that feed on them. The chemical functionality reviewed includes isocyanides, isothiocyanates, formamides, thiocyanates, isocyanates, and dichloroimines. The literature through mid 2003 is reviewed and there are 143 citations.
 Mary J. Garson | Mary Garson obtained a PhD in organic chemistry under Professor Jim Staunton at the University of Cambridge in 1977. After postdoctoral work in Italy, followed by two years in the pharmaceutical industry, she migrated to Australia in 1983 as a Queen Elizabeth II Research Fellow at James Cook University of North Queensland where she began to study isocyanide biosynthesis. She has held academic positions at The University of Wollongong and (currently) The University of Queensland, where she is a Reader in Organic Chemistry. Additionally she is Chair of the Rio Tinto Australian Science Olympiads. Her interest lies in determining how the structures of marine natural products relate to their biological functions, and therefore encompasses biosynthesis and ecology. She is a keen scuba diver, especially in the warm waters of Queensland. |
 Jamie S. Simpson | Jamie Simpson received his BSc (Hons) in Chemistry from The University of Queensland in 1995, and then obtained his PhD in 2000 under the supervision of Associate Professor Mary Garson. After a short term appointment as Lecturer in Chemistry at The University of New England, he moved to his current position as a Research Fellow at the Research School of Chemistry at The Australian National University, in the laboratory of Professor Chris Easton. His research interests are in the general area of biological and organic chemistry, including natural products chemistry, biosynthesis and enzyme–small molecule interactions. |
1 Introduction
Marine invertebrates and the animals that feed on them contain an abundance of unique bioactive nitrogenous compounds, among which are the terpene isocyanides.1–3 There has been interest in the biosynthesis of terpene isocyanide metabolites, notably in the origin of the isocyanide group, ever since the first report of their isolation in 1973.4 As well as biosynthesis, recent attention has also focused on the ecological roles of this extraordinary group of metabolites. John Faulkner wrote in 1977 that he “could see no more interesting study among the marine natural products”.5 In view of his well-documented interest in linking marine natural products to their true biological roles, it seems highly appropriate to review current knowledge on the chemistry and biosynthesis of this intriguing class of marine natural product, and to place this chemistry in a biological context.This review updates the coverage given to marine isocyanides in the 1988 Natural Product Reports article of Edenborough and Herbert6 but expands the topic to include discussion of related functionality, notably marine natural products containing isothiocyanate, thiocyanate, isocyanate and dichloroimine substituents, and also the structurally-related formamides. Marine isocyanide biosynthesis was last reviewed in 1990 by Chang and Scheuer,7 and so this topic is given detailed coverage in this article.
2 Marine isocyanides and related structures
Two comprehensive reviews survey naturally occurring isocyano/isothiocyanato natural products and related compounds from both marine and terrestrial sources up to 1993 and 1999 respectively.1,2 In view of this extensive coverage, the current compilation only reviews new natural products reported since mid 1999. Many terpene isocyanides isolated from natural sources are accompanied by the corresponding isothiocyanate and formamide substituted metabolites, hence these three functional groups are best considered collectively.2.1 Marine isocyanides, isothiocyanates and formamides
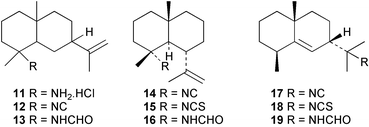
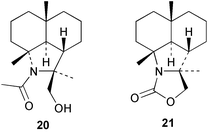
The Caribbean sponge Axinyssa ambrosia has yielded a novel amine hydrochloride 11 with an eudesmane skeleton, together with the corresponding isocyanide 12 and formamide 13.17 Also isolated were the known 4α-substituted NC/NCS/NHCHO triad of gorgon-11-ene 14–1618 and the 11-substituted NC/NCS/NHCHO triad of 7β-H-eudesm-5-ene 17–19.19 Compound 11 was cytotoxic against P388, A-549 (human lung carcinoma) and HT-29 (human colon carcinoma) cell lines. The sponge Axinyssa terpnis also contains 4α-isocyanogorgon-11-ene (14) and its formamido analogue 16.20 Compounds 14 and 16 were cytotoxic to murine leukaemia P388 cells with LC50's of 1.2 and 8.3 µg mL−1 respectively, whereas only isocyanide 14 showed activity in a brine shrimp assay. Attempted oxidation of 14 or 16 with MCPBA gave the novel cyclisation products 20 and 21 respectively.20
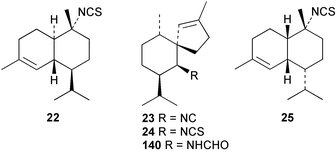
10-Isothiocyanato-4-cadinene (22) has been independently described by two groups, with some differences in 1H and 13C NMR data. It was first reported in 2000 from an Australian sample of Acanthella cavernosa,21 and subsequently in 2003 from the Fijian Phakellia carduus and its nudibranch prey Phyllidiella pustulosa together with the commonly encountered sesquiterpene axisonitrile-3 23.22 Despite the many reports of its isolation from sponges, the absolute stereochemistry of axisonitrile-3 continues to be a source of confusion in the literature. The correct configuration for (+)-axisonitrile-3, deduced by synthesis of the optical antipode from (+)-dihydrocarvone,23 is shown in structure 23.3,6 Isothiocyanate 22 has moderate antimalarial activity,22 as does axisothiocyanate-3 (24), whereas axisonitrile-3 shows potent dose-dependent antimalarial activity with cultured Plasmodium falciparum,24 and has antimycobacterial activity.25 These data support the suggestion that the isocyano group is important for antiplasmodial activity.24 10-Isothiocyanato-4-amorphene (25), isolated from a Halichondria sp.,26 has been reisolated and subjected to a range of biological, including antimalarial, insecticidal, herbicidal and fungicidal assays, but did not show useful activity.27,28
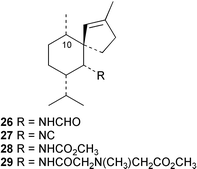
A Japanese specimen of Geodia exigua contains some novel spiroaxane metabolites. The spirocyclic exiguamide (26) was isolated together with 10-epi-axisonitrile-3 (27) and two novel analogues exicarbamate (28) and exigurin (29), have also been isolated.29,30 The relative stereochemistry of exiguamide was solved by NMR study and by an X-ray analysis,29 while the absolute stereochemistry for 26–29 was determined by applying the modified Mosher's method on an amine derivative.30 This revealed that these compounds are the C-10 epimers of (−)-axisonitrile-3 (which has not to our knowledge been reported from natural sources). The methyl carbamate 28 and amide 29, which was isolated in trace amounts, both represent nitrogenous derivatives possessing unusual functionality. These compounds are of obvious biosynthetic interest since their occurrence suggests a precursor role for isocyanates or amines in this sponge. 10-Epi-axisonitrile-3 (27) was previously isolated from the nudibranch Phyllidiella pustulosa.11
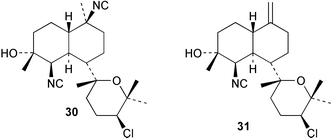
The functionalised decalin core of kalihinol A (30) has been synthesised by an intramolecular Diels–Alder cyclisation.33 Two recent studies provide details of the absolute stereochemistry of kalihinane metabolites. The absolute configuration of kalihinol A (30) has been determined by application of the CD exciton chirality method to a bis-p-bromobenzamide derivative prepared from the corresponding bisamine.34 This work, which is the first report of absolute stereochemistry in the kalihinol tetrahydropyran series, confirms a provisional configurational assignment made on the basis of CD measurements for a ketone derived from kalihinol Y (31).35 The absolute stereochemistry of kalihinene X (32) has been confirmed by total synthesis involving regioselective coupling of an alkyl sulfone with an epoxyalcohol followed by an intramolecular Diels–Alder reaction,36 and corresponds to that determined for kalihinol A except for the epimeric C-1. In view of the variable or zero optical rotations reported in the literature for other kalihinane metabolites, a full stereochemical picture is not yet apparent for this important class of marine diterpenes. Hopefully future studies will determine if other members of the kalihinol/kalihinene series share the same stereochemistry as the above two compounds. To avoid confusion when consulting the literature, kalihinane metabolites with unknown absolute stereochemistry have been presented in this review as drawn in the original articles.
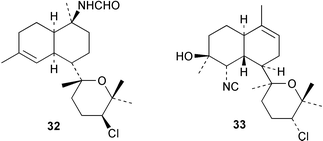
Three new kalihinols Δ9-kalihinol Y (33), 10-epi-kalihinol I (34) and 5,10-bisisothiocyanatokalihinol G (35) have been isolated from an Okinawan Acanthella sp. along with kalihinol A (30), kalihinene (36), and 6-hydroxykalihinene (37). All six compounds were active when tested against P. falciparum, with kalihinol A showing potent and selective antimalarial activity.37
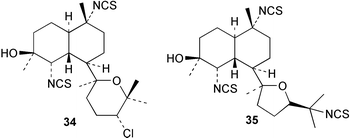
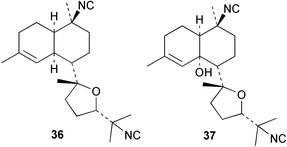
An Australian Acanthella cavernosa was reported to contain two epoxy-substituted isocyanobifloradienes 38 and 39, together with 8-hydroxyisokalihinol F (40), and the known metabolites isokalihinol F (41), 1-epi-kalihinene (42) and kalihipyran (43). Relative stereochemistry was deduced by NOESY data; in particular, a trans ring configuration was deduced for these compounds by the differing sets of NOESY correlations shown by H-1 and H-6.21 In the bifloradienes 38 and 39, the appearance of H-5 (singlet) and H-6 (doublet) are also consistent with trans geometry.38 A new bifloradiene hydrocarbon 44 has been isolated from Cribochalina sp.,39 and is reported to have different stereochemistry to bifloradiene 45 described earlier from A. cavernosa.40 Despite the differing ring junction stereochemistry shown, the two compounds have matching NMR data for the bicyclic ring system. The [α]D values for the two compounds are also quite similar, +51.4° for 44 and +64° for 45. Evidence supporting the suggested C-11 configuration in both 44 and 45 is lacking. In sponges such as A. cavernosa, compounds of this type may represent metabolic endpoints unable to proceed to functionalised diterpenes. The absence of the C-11, C-12 double bond prevents further cyclisation to the tetrahydrofuran or tetrahydropyran ring systems found in the kalihinols. (−)-Axinyssene (46) from Axinyssa sp. is a diterpene hydrocarbon showing a novel carbon skeleton, and with an IC50 value of 16.9 µg mL−1 against a HL-60 cell line. The absolute configuration at C-4 was determined to be S by synthesis from (−)-limonene and geraniol,41 but the structure 46 depicted in the article, and therefore drawn here, has R stereochemistry.
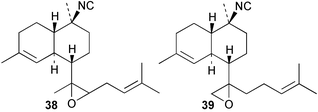
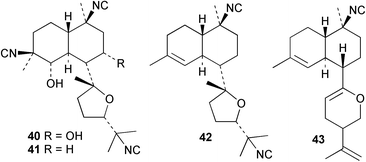
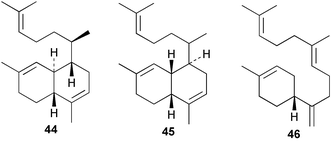
Cis and trans-fused bifloradiene hydrocarbons such as 47 and 48 can be proposed as intermediates in the biosynthesis of kalihinenes (e.g.32, 36, 37, 42), kalihinols (e.g.30, 31, 33–35, 40–41, 49) and related metabolites such as 38, 39, 44 and 45. Scheme 1 shows a detailed biosynthetic pathway for biosynthesis of selected isocyanide metabolites based on earlier proposals of Rodriguez42 and Chang and Scheuer.7 The confirmed configuration of kalihinol A (30) provides the basis for the stereochemistry shown in this Scheme. A similar route (not shown) can explain the biosynthetic origins of isothiocyanato metabolites. The isocyano substituents in the bicyclic nucleus could be introduced by attack of cyanide ion on a carbocation (or equivalent) generated by protonation of a precursor double bond. Formation of the furan ring or pyran ring could be achieved by (i) epoxidation of the C-14, C-15 double bond followed by (ii) attack of cyanide at either C14 (path a), ultimately generating pyrans (e.g.30, 32), or at C-15 (path b), ultimately generating the furan series of metabolites (e.g.36, 49). The kalihinol (e.g.49) or isokalihinol (e.g.41) series of metabolites mechanistically result from (i) epoxidation of the C-4, C-5 double bond followed by (ii) attack of cyanide at either C4 (isokalihinols) or at C-5 (kalihinols). The two series of metabolites thus differ in the relative positioning of the C-4 and C-5 hydroxyl and isocyano (isothiocyanato) substituents. The metabolites 38 and 39 are of biosynthetic significance for two reasons; their isolation supports the proposed role of bicyclic intermediates (e.g.47 and 48), and further confirms the ability of A. cavernosa to epoxidise double bonds. It is noteworthy that epoxide 39 is mechanistically the logical precursor to kalihipyran 43 (Scheme 2),21 in contrast to the earlier biosynthetic route proposed by Rodriguez.42
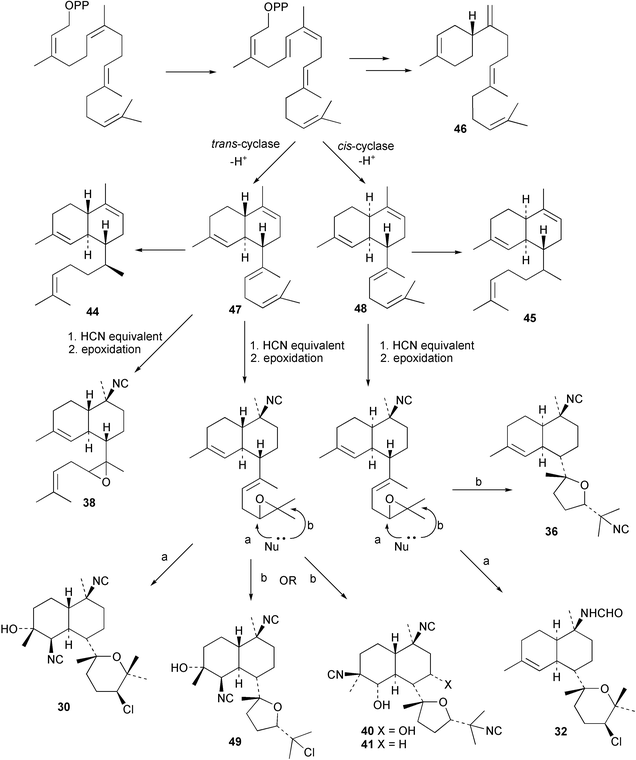 | ||
| Scheme 1 Proposed biosynthetic pathway leading to the kalihinols (revised from Rodriguez et al.).42 | ||
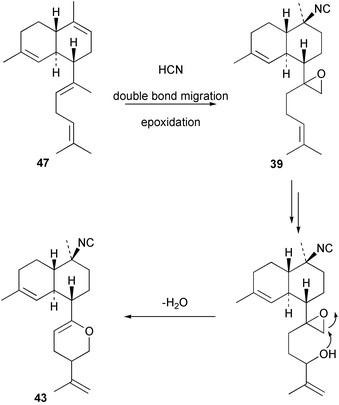 | ||
| Scheme 2 Proposed biosynthetic pathway leading to the kalihipyrans. | ||
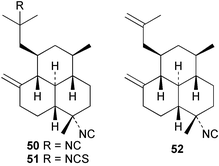
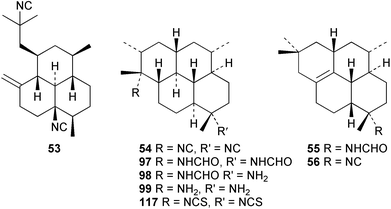
Three new isocyano functionalised diterpenes, 50–52, each with an amphilectene skeleton have been isolated from Cribochalina sp. alongside the above mentioned bifloradiene 44 and the known isocyanide 53,39 previously isolated by Wratten from Hymeniacidon sp.43 The absolute configuration proposed for 53 was deduced by Mosher ester analysis on an alcohol prepared by reductive ozonolysis,39 and is opposite to that demonstrated for the related isocycloamphilectane 7,20-diisocyanoadociane (54) by total synthesis44 and by X-ray analysis of a derivative.45 A new formamido-substituted cycloamphilectene metabolite 55,46 which is related to the isocyanide 56 previously isolated from a Palauan Halichondria sp,47 has been reported from Axinella sp. from Vanuatu. The structure of 55 was solved by spectroscopic and X-ray studies.46
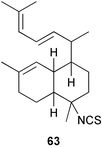
Scheme 3 shows plausible biosynthetic pathways leading to the known amphilectene/cycloamphilectene metabolites 54, 57–61, based on cyclisation and functionalisation of the bicyclic intermediate 47. This suggested intermediate is also a central intermediate in the proposed pathway to kalihinanes (Scheme 1). Evidence supporting the proposed role of 47 in amphilectane biosynthesis is provided by the isolation of bifloradiene metabolites from two sponges. Compound 62 has been reported from Cymbastela hooperi,48 a rich source of the amphilectane class of marine diterpenes. A sponge of the family Adociae produces 63,49 which is known to differ in stereochemistry to 62.48 While no relative stereochemistry was assigned to compound 63 by the authors, the NMR data provided is suggestive of cis stereochemistry when compared to literature data for kalihinene and epi-kalihinene metabolites.21,38 Standard terpene cyclisations could convert 47 into 58–61, while the biosynthetic steps leading to 54 and 57 are more complex, presumably involving alkyl migrations.
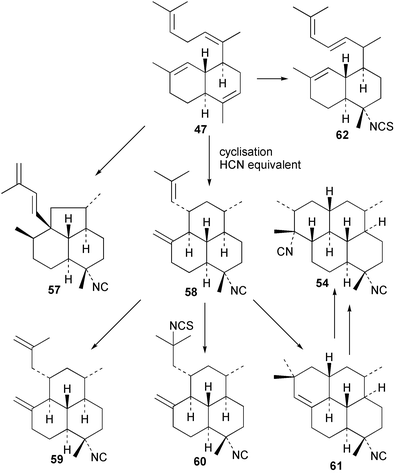 | ||
| Scheme 3 Proposed biosynthetic pathway leading to the amphilectane diterpene isocyanides. | ||
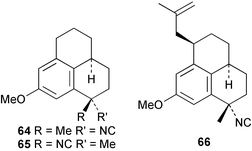
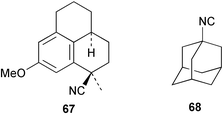
As mentioned previously, several marine sesqui- and diterpenes have been reported to show useful antimalarial activity.24,50,51 One of the most potent marine antimalarials is 7,20-diisocyanoadociane 54 whose molecular mechanism of action has been investigated. The potency of isocyanides such as 54 as antimalarials is related to their ability to bind iron porphyrins and prevent their conversion to β-hematin; the buildup of toxic heme moieties then leads to destruction of the malaria parasite membranes.52 The pronounced antimalarial activity exhibited by marine-derived isocyanides prompted Schwarz et al. to synthesise the tricyclic isocyanides 64–66, and a related nitrile 67, using arene tricarbonyl chemistry and evaluate them for antimalarial potential. All of the isocyanides were active, although less potent than the naturally-occurring marine analogues, whereas the nitrile was inactive.53 Singh et al., have evaluated a range of simple isocyanides for their antimalarial activity and found that adamantyl isocyanide 68 showed some potency.54
2.2 Thiocyanates and isocyanates
According to Chang,2 five thiocyanato-substituted terpenes have been reported from marine sponges51,55–57 or the molluscs that feed on them.11,58 The syntheses of two of these have now been reported. 4-Thiocyanatoneopupukeanane (69) and both epimers (70, 71) of 2-thiocyanatoneopupukeanane (which occurs naturally as 7156) have been prepared from (R)-carvone using rhodium carbenoid C–H insertion chemistry to prepare the isotwistane ring system.59,60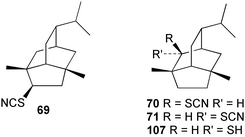
As reviewed by Chang,2 five marine isocyanates have been reported in the marine literature. To our knowledge, no new thiocyanates or isocyanates have been reported since 1999.
2.3 Dichloroimines
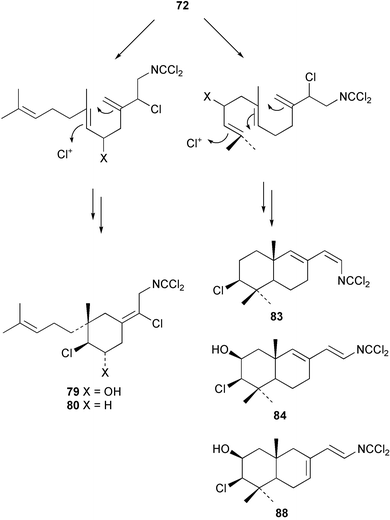
In general, metabolites containing a dichloroimine (=carbonimidic dichloride) group are rare; to date, this functionality appears exclusively in marine sesquiterpenoid metabolites. Since the first report of their isolation by the Faulkner research group,61 more than a dozen sesquiterpenes (72–88) with this uncommon functionality have been reported from sponges and molluscs. Metabolites showing both linear and cyclic carbon skeletons have been described.2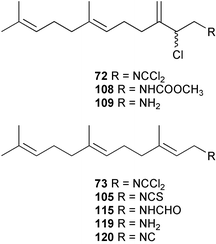
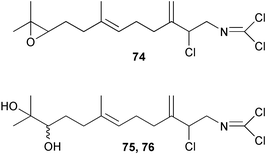
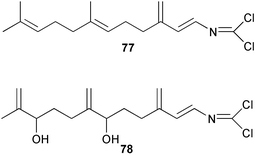
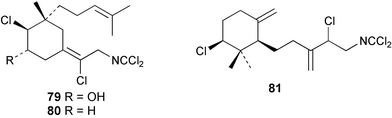
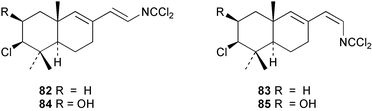
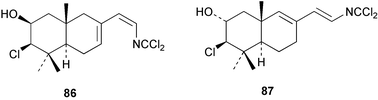
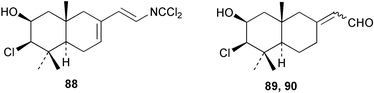
Two new linear dichloroimine metabolites, named as ulosins A (77) and B (78) have been isolated as unstable diene dichloroimines from Ulosa spongia,62 along with the known stylotellane B (72), and the bicyclic isoreticulidin B (84) and reticulidin B (88). Ulosin A (77) has also been isolated from an Okinawan collection of Stylotella aurantium, with some minor differences in 13C data reported.63S. aurantium also contained the novel compounds 80 and 81, and the bicyclic 82, as well as two unusual aldehydes, 89 and 90, which may be either decomposition or catabolic products of reticulidin-type metabolites.63,64 Interestingly this collection also contained reticulidins A (87) and B (88), which had earlier been isolated from the mollusc Reticulidia fungia, also collected in Okinawa,65 suggesting S. aurantium as the dietary source of these compounds in the mollusc. The absolute stereochemistry of reticulidin A (87) was determined by a modified Mosher's ester procedure, however this approach was not successful with reticulidin B.63 The isolation of 79, 84, and 88 from an Australian collection of S. aurantium prompted the authors to report complete and revised NMR data for these known compounds.66 Metabolites 81, 82, 87, 88, 89, and 90 are cytotoxic to a range of cell lines,63,65,67 with some of these compounds (81, 82, 89, and 90) being the subject of a patent.68 Curiously, while Australian and Japanese specimens of S. aurantium contain dichloroimines, other specimens from the Indo-Pacific are reported to contain sesquiterpene isocyanides, cyclopeptides or chlorinated guanidines.69–79 A taxonomic reinvestigation of dichloroimine-containing sponges is desirable.
2.4 Miscellaneous compounds of non-terpenoid metabolism
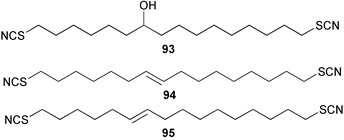
The thiocyanate psammaplin B (91) has been isolated as a minor metabolite of the Indo Pacific sponge Pseudoceratina purpurea and shows modest activity as a histone decarboxylase inhibitor.80 Fasicularin (92) is a novel tricyclic alkaloid isolated from the Micronesian ascidian Nephteis fasicularis, whose structure is analogous to that of cylindricines F–H isolated from the ascidian Clavelina cylindrica,81,82 all of which contain a thiocyanato substituent. The co-occurrence of cylindricine I containing an isothiocyanato group,82 supports the role of the ambident thiocyanate in the biosynthesis of this suite of metabolites. Fasicularin, which is active in a DNA damaging assay, has been the subject of two total syntheses.83,84 Both approaches used Diels–Alder chemistry to prepare the trans-perhydroisoquinoline ring system. In one synthesis, the stereocontrolled hetero Diels–Alder reaction involved an N-acylnitroso derivative,83 while the other used an intermolecular cycloaddition of a substituted acrolein with the dioxolane ketal of a 1,3-diene-7-one.84 Three novel thiocyanatin metabolites 93–95 have been isolated from a Western Australian sponge Oceanapia sp. Racemic thiocyanatin A (93) was synthesised in seven steps from 8-bromooctanoic acid, as were a mixture of thiocyanatins B and C (94) and (95). The natural and synthetic 93 showed nematocidal activity against Haemonchus contortus while no in vitro nematocidal activity was found for either natural or synthetic 94/95.85 The structures of these compounds are reminiscent of a group of long chain aliphatic α,ω–bisisothiocyanates isolated from a Fijian Pseudaxinyssa sp.86 A plausible role for the ambident nucleophile thiocyanate in the biosynthesis of these compounds from fatty acid or polyketide precursors can be envisaged.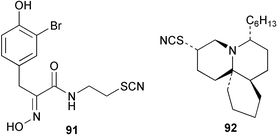
3 Biosynthesis of marine isocyanides and related compounds
Two intriguing aspects of marine terpene isocyanide biosynthesis are the origin of the terpene skeleton and the origin of the attached nitrogenous functional group. Studies on these topics have been undertaken in both sponges and molluscs.3.1 Protocols for biosynthetic study
Although in situ experiments are possible,87 precursor incorporation studies using marine sponges are generally performed using healthy specimens maintained in aquaria.45,88,89 After collection, sponges are allowed to equilibrate in aquaria with flowing fresh seawater, before being incubated (ca. 12 h) with precursor in small glass jars (ca. 200 mL). The sponges are then returned to aquaria for an interval (typically 10–20 days depending on the health of the specimens) to allow the precursor to be incorporated into natural products which are then isolated using standard methods. In experiments using radioactive precursors, the isolated products must be rigorously purified to constant radioactivity, and the sites of labelling determined by chemical degradation.88,90,91In situ experiments have been undertaken by embedding the precursor into animal tissue using gelatin capsules,87 or by injection of precursor into the live animal.92,93 Experiments on the biosynthesis of mollusc metabolites are usually undertaken by injection of the precursor into the mollusc.94 In one study, the mollusc was allowed to feed on sponges containing radiolabelled metabolites resulting from incorporation of a radiolabelled precursor.89
3.2 Biosynthesis in marine sponges
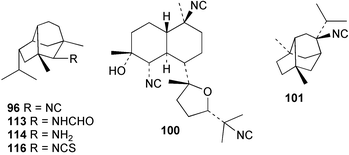
Research by Garson was the first to show that the isocyanide groups of diisocyanoadociane (54) are derived from inorganic cyanide through incorporation studies with the Great Barrier Reef sponge Amphimedon terpenensis.88 The [14C]-label was proven to be specifically associated with the isocyanide carbons through stepwise hydrolytic degradation to the corresponding formamide 97, and then to amines 98 and 99. The radioactive label was shown to be equally distributed between the two isocyano positions, and higher levels of incorporation were obtained after 34 days (1.83%) compared to 18 days (0.46%).88 Subsequently, studies by Karuso and Scheuer confirmed that [14C]-cyanide is a precursor for 2-isocyanopupukeanane (96) in a Hawaiian Ciocalypta sp. and the diterpene kalihinol F (100) in Acanthella sp. from Guam.93 The precursors were encapsulated in lipid vesicles and injected into the sponges that were then maintained in their natural habitats for 3–5 weeks before collection and extraction. Incorporation levels of 1.5–1.8% were achieved. The radiolabelled 100 was converted via the trisamine to a trisbenzylurea derivative which was not radioactive, thereby establishing that the radioactivity was associated with the isocyanide carbons. The cyanide incorporation was shown to be an enzymatic process since there was no incorporation of [14C]-cyanide in the absence of sponge tissue. These researchers next explored the incorporation of [13C, 15N]-cyanide into a specimen of Ciocalypta sp. that contained 9-isocyanoneopupukeanane (101). A 0.5 g sample of potassium [13C, 15N]-cyanide was provided to the sponge in a series of five injections spread out over 9 weeks. 13C NMR analysis revealed a 1.46% enrichment of the isocyano carbon compared to unlabelled material. Difference spectroscopy gave good cancellation of all signals except for the isocyano carbon which remained as a doublet (J = 5.9 Hz), 0.037 ppm upfield of the unlabelled resonance. These results show that both the carbon and the nitrogen atoms of the cyanide moiety are incorporated intact into isocyanide 101 in Ciocalypta sp.93
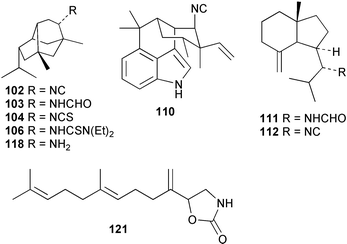
The utilisation of inorganic cyanide for terpene isocyanide biosynthesis has been studied in two other Australian sponges. Provision of [14C]-cyanide to A. cavernosa for 16 days gave 0.15–0.39% incorporations into the major sesquiterpene metabolite axisonitrile-3 (23).89 These incorporation levels are comparable to those obtained with A. terpenensis,88Ciocalypta sp. and the Guam Acanthella sp.93 In contrast, the incorporation of cyanide ion into 9-isocyanopupukeanane (102) in Axinyssa n.sp. in two separate experiments with incorporation periods of 16 or 27 days was found to be very low, despite the significant recovery of metabolite (> 10 mg per experiment).91,97 Typically, a radioactivity content of less than 100 dpm mg−1 (< 0.001%) was obtained following incorporation of up to 100 µCi of precursor. Conversion of the isolated samples of isocyanide 102 into the corresponding formamide 103 occurred with full retention of radioactivity and established that the labelling, although low, was genuine. It is interesting to reflect that if incorporation levels of this magnitude had been obtained in the initial biosynthetic experiments carried out with A. terpenensis by Garson, the role of inorganic cyanide in marine isocyanide biosynthesis may not have been appreciated.
The first studies on the biosynthesis of marine isothiocyanates involved Australian specimens of A. cavernosa and established that thiocyanate ion was a precursor to the isothiocyanate moiety in axisothiocyanate-3 (24). The incorporation levels obtained in experiments of 11 days duration on two sponge specimens were low (0.3%) which in this instance reflected the low recovery (< 1 mg) of the isothiocyanate 24. Inorganic cyanide was also a precursor to the isothiocyanate moiety in this sponge, suggesting a facile conversion of cyanide to thiocyanate had occurred.89 The enzyme rhodanese, widespread in Nature, detoxifies cyanide through conversion to thiocyanate,98 but it is not yet known if this enzyme, or its equivalent, is present in marine sponges such as A. cavernosa.
Subsequently, both cyanide and thiocyanate ion have been incorporated into the isothiocyanato groups of 9-isothiocyanatopupukeanane (104) by Axinyssa n.sp.,91,97 and farnesyl isothiocyanate (105) in S. aurantium.90,97 Incorporation levels of 0.3–1.14% were obtained for isothiocyanate 104 in Axinyssa n.sp., in complete contrast to the low incorporations obtained for the isocyano co-metabolite 102. The radioactive 9-isothiocyanatopupukeanane (104) samples from the cyanide and thiocyanate incubations were converted to the N,N-diethylthiourea (106) products that retained radioactivity. Incubation of cyanide and thiocyanate with S. aurantium gave farnesyl isothiocyanate (105) with variable incorporations (0.01–2.0%), depending on the amount of metabolite isolated. Attempts at hydrolytic degradation of these thiocyanates were unsuccessful, but it was assumed that the 14C label was associated with the isothiocyanate functionality by analogy with the earlier research on isocyanides.
In [14C]-thiocyanate incorporation experiments with A. cavernosa89 and Axinyssa n.sp.,91 the co-occurring isocyanides axisonitrile-3 (23) and 9-isocyanopupukeanane (102) were also labelled. Incorporation levels were 0.3–0.6% and < 0.01% for the isocyanides 23 and 102 respectively. The low values obtained for metabolite 102 were comparable to those obtained in cyanide incorporations.91,97 The role of thiocyanate as a precursor to diisocyanoadociane (54) in A. terpenensis was confirmed by incorporation of [14C]-thiocyanate to give radiolabelled diisocyanoadociane, with the specificity of labelling confirmed by stepwise degradation.99 Overall, the results imply that marine sponges such as A. cavernosa, Axinyssa n.sp. and A. terpenensis can convert thiocyanate to cyanide. A number of enzymes are known to convert thiocyanate into cyanide ion.100,101
The biosynthetic origin of thiocyanates has generated interest. Scheuer and co-workers57 suggested the cyanation of a thiol, which appears to be a plausible pathway to amino acid-derived metabolites such as psammaplin B (91). In marine sponges, terpene thiocyanates often co-occur with isothiocyanates, and here the involvement of the ambident nucleophile thiocyanate is mechanistically reasonable.55,56 Experimental proof was obtained when Axinyssa n.sp. was shown to convert [14C]-thiocyanate into 2-thiocyanatoneopupukeanane (71).91 LiAlH4 reduction of 71 gave the unlabelled thiol 107, confirming that the radioactive label was exclusively associated with the thiocyanato moiety. In a second experiment, [14C]-cyanide was also incorporated into the thiocyanato group of 71.91,97 This result gives stronger support to the proposal that sponges convert inorganic cyanide into inorganic thiocyanate (Scheme 4). It is difficult to envisage the addition of cyanide to a terpene precursor, followed by insertion of sulfur to give a thiocyanate. Sulfur addition to give an isothiocyanate followed by isomerisation remains a possibility, however in chemical reactions of this type the equilibrium usually favours an isothiocyanate over a thiocyanate.102 In both the cyanide and thiocyanate experiments, the specific activity of the thiocyanate 71 decreased significantly when extended incorporation periods were used.91,97
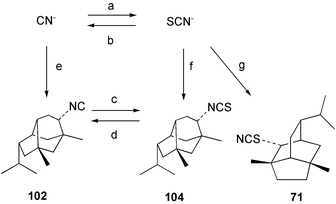 | ||
| Scheme 4 Biosynthetic pathways in Axinyssa n.sp. | ||
Faulkner and Wratten first speculated that the biosynthetic origin of the dichloroimine functional group might be linked to that of the corresponding isocyanide61 or isothiocyanate.103 Farnesyl isothiocyanate (105) has been isolated as a co-metabolite of sesquiterpene dichloroimine metabolites in sponges such as P. pitys and S. aurantium.90,103 Experiments then showed that S. aurantium converts [14C]-cyanide into stylotellane A (73) and stylotellane B (72). The labelled stylotellane B was degraded (H3PO4; 0.1 N in MeOH) to give the labelled carbamate 108 together with unlabelled amine 109, showing that cyanide is specifically incorporated into the dichloroimine functional group. A parallel set of experiments was conducted using [14C]-thiocyanate as precursor and also gave radiolabelled stylotellanes A and B. The lack of sufficient radiolabelled metabolite prevented chemical degradation to establish the sites of labelling, however it was anticipated that the [14C] label from thiocyanate was incorporated specifically into the dichloroimine carbon atom. Scheme 5 presents plausible mechanisms through which cyanide or thiocyanate ion might be implicated in dichloroimine biosynthesis.90 Chlorination of isocyanides or isothiocyanates is known to produce dichloroimines under laboratory conditions.104 A similar chlorination may occur enzmatically to give the dichloroimine metabolites. Halogenating enzymes are widespread in marine systems.105
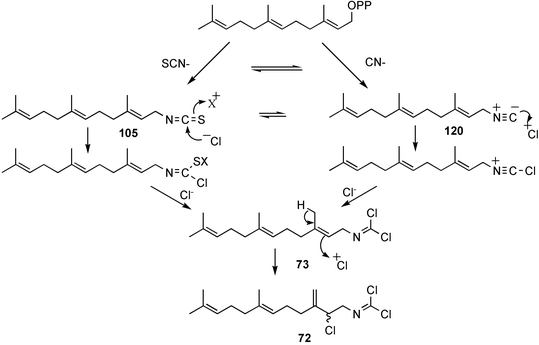 | ||
| Scheme 5 Biosynthetic scheme leading to the stylotellanes. | ||
Additional biosynthetic experiments were performed with S. aurantium to investigate how the incorporation levels and specific activities changed over time. A comparison of the incorporation results from a number of experiments revealed an interesting trend. The specific activity and incorporation levels decreased for farnesyl isothiocyanate (105) and increased for stylotellane B (72) over time, consistent with the rapid formation of isothiocyanate 105 from the precursor, while the dichloroimine 72 is formed more slowly. This is consistent with the isothiocyanate being converted to the dichloroimine over the longer incorporation period.97 A possible role for farnesyl isothiocyanate in dichloroimine biosynthesis is shown in Scheme 5.
Overall, the inorganic precursor experiments carried out with A. terpenensis, A. cavernosa, Axinyssa n.sp., and S. aurantium all support precursor roles for both cyanide and thiocyanate ion in the biosynthesis of the various isocyano, isothiocyanato, thiocyanato and dichloroimine metabolites. Further, an interconversion of cyanide and thiocyanate ion can occur in these sponges. From a biological perspective, the results are intriguing given the proven role of cyanide as a cytochrome oxidase inhibitor.106 Given that the concentrations of these ions in sea water are negligible,107 the biochemical origin of the cyanide/thiocyanate utilised by these sponges is of interest. Attempts to probe single carbon precursor roles for amino acids such as alanine, arginine, glycine or leucine in diisocyanoadociane (54) biosynthesis did not yield labelled metabolite.45
It is possible that microbial symbionts present in these marine sponges may play a role in cyanide production. Microbial sources of cyanide have been documented.106 An interesting biosynthetic comparison to the marine isocyanides may be found in the terrestrial metabolite hapalindole A (110) which is an isocyano-containing metabolite of amino acid/terpenoid origin produced by the cyanophyte Hapalosiphon fontinalis. Incorporation experiments with radiolabelled formate, glycine, methionine and serine have established that the isocyanide group originates from C1-tetrahydrofolate metabolism in this microorganism. Interestingly, a 0.16% incorporation of [14C]-cyanide was also reported.108
As mentioned in Section 3.2.1, the first proposals for marine isocyanide biosynthesis focused on whether formylation of an amine, and dehydration of the resulting N-formyl compound, might ultimately provide a terpene isocyanide. Although this biosynthetic proposal was eventually negated by a formate labelling study, earlier advanced precursor studies by Iengo et al. had suggested that formamides do not act as biogenetic precursors to isocyanides.95 [14C]-Labelled axamide-1 (111), prepared by formylation of the corresponding amine, was provided to the Mediterranean sponge Axinella cannabina for a five day period. The isolated axisonitrile-1 (112) was not labelled, although the precursor was taken up and metabolised by the sponge as evidenced by incorporation of label into a free fatty acid fraction. This result was followed by an incorporation experiment carried out by Hagadone et al. with Hymeniacidon sp. (syn Ciocalypta sp.) to evaluate the origin of the isocyano group in 2-isocyanopupukeanane (96). [13C]-Labelled samples of the formamide (113), synthesised from the corresponding amine (114) and H13CO2H, were provided to the sponge in an in situ feeding experiment. The biosynthetic data, obtained using the relatively insensitive technique of mass spectrometric detection, suggested that the formamide is not a biosynthetic precursor.87 Recently, Brust and Garson have confirmed that formamide intermediates are not involved in dichloroimine biosynthesis. When [14C] labelled farnesyl formamide (115) was provided to S. aurantium, the precursor was taken up by the sponge, but no conversion into stylotellanes A (73) and B (72) occurred.96
The interconversion of isocyanides and isothiocyanates has been studied in several sponge systems. Incorporation experiments were carried out by Hagadone et al. in their study of Hymeniacidon sp. (syn Ciocalypta sp.). They synthesised [13C]-labelled samples of 2-isocyanopupukeanane (96) by dehydration of formamide 113 with POCl3, then converted a portion of this isocyanide to the isothiocyanate 116 (S, heat at 110 °C). The in situ labelling study suggested that 2-isocyanopupukeanane (96) was a precursor to the corresponding isothiocyanate 116, but the reverse isothiocyanate to isocyanide conversion was not detected. However they reported very low incorporation levels that were detected using mass spectrometry, and hence these results are unconvincing.87
Isocyanide–isothiocyanate interconversions have been more effectively probed in two other marine sponges, A.terpenensis97,99 and Axinyssa n.sp.97,111 In the A. terpenensis study, the bisamine 99 prepared by acid hydrolysis of 54 was converted to the [14C]-labelled formamide 97 using a mixed formic-pivalic anhydride. Dehydration (TsCl, pyr.) then gave [14C]2-diisocyanoadociane (54), and sulfur treatment gave [14C]2–bisisothiocyanatodociane 117 in modest overall radiochemical yield.97,99 Initially, [14C]2-bisisothiocyanate 117 was provided in duplicate to small pieces of A. terpenensis. After 19 days incorporation, the diisocyanoadociane (54) isolated from each specimen was found to contain a low level of radioactivity. One sample of 54 was degraded to the formamide 97, which retained the radioactivity, while the other sample was hydrolysed directly to the bisamine 99 that contained 9.1% of the activity of the parent diisocyanide. The higher than expected 14C content of this sample of 99 was attributed to side products of the hydrolysis reaction, and the difficulty of purification.99 The data from these initial advanced precursor studies was complicated by concerns about the radiochemical purity of the precursor supplied, given that it had been prepared from radioactive 54. Subsequently, an incorporation experiment was conducted on A. terpenensis using a sample of bisisothiocyanate 117 that had been treated with unlabelled 54 to remove any traces of residual [14C]2-54. The isolated diisocyanoadociane had a radioactivity content of 4 dpm mg−1 above background, and was essentially non radioactive. Thus there was no incorporation of bisisothiocyanate 117 into diisocyanoadociane 54 in this experiment.97 The bisisothiocyanate 117 has never been isolated from A. terpenensis, or from the closely-related C. hooperi.48
A second study involved Axinyssa n.sp., the sponge that contains the 9–isocyanopupukeanane (102) and 9-isothiocyanatopupukeanane (103) pair of metabolites.111 [14C]-Isocyanide 102 and [14C]-isothiocyanate 104 were prepared using a similar [14C]-formic-pivalic anhydride procedure, utilizing the amine 118, available by 6 M HCl hydrolysis of sponge-derived 102. The resulting sample of [14C]-104 was mixed with unlabelled 102, and the two individual components separated. Two cycles of this purification protocol were used to confirm the radiochemical purity of the recovered [14C]-9-isothiocyanatopupukeanane (104) for use in incorporations. An incorporation experiment with Axinyssa n.sp. using [14C]-9-isocyanopupukeanane (102) resulted in radioactive 9-isothiocyanatopupukeanane (104), which was converted to the radioactive thiourea derivative 106 (Et2NH reflux). In a second experiment, [14C]-9-isothiocyanatopupukeanane (104) was supplied to the sponge. The sample of 9-isocyanopupukeanane (102) isolated from this incorporation experiment was significantly radioactive, as was the formamide 103 resulting from glacial acetic acid treatment. These results show the conversion of isocyanides to isothiocyanates and also of isothiocyanates to isocyanides in Axinyssa n.sp. The high incorporation levels obtained do not suggest that metabolic degradation and re-utilisation of labelled breakdown products via general metabolism had occurred.
The possibility that these sponge interconversions could occur without the action of enzymes was considered.97 Some evidence supporting the role of enzymes was available from the synthetic procedure used (no transfer of radiolabel was observed on mixing [14C]-isothiocyanate 104 with unlabelled isocyanide 102, and from the negative result obtained for the bisisothiocyanate 117 to isocyanide 54 incorporation in A. terpenensis. Had any chemical interchange of label occurred in either of these experiments, it would have resulted in labelled isocyanide. These observations thus suggest that the non-enzymatic conversion of isothiocyanates to isocyanides either does not occur, or occurs only at a very slow rate under incorporation conditions.
In Axinyssa n.sp., the major biosynthetic route (Scheme 4) appears to be from cyanide to thiocyanate, followed by incorporation into the thiocyanate 71 or isothiocyanate 104 and subsequent conversion of the isothiocyanate into the isocyanide 102. If these steps (Scheme 4, reactions a,f,g) occurred at a slow rate, then isocyanide 102 (formed via d) would be labelled at a lower level than the isothiocyanate 104, as was observed experimentally for cyanide and thiocyanate incorporations. The thiocyanate incorporations are consistent with the proposed biosynthetic route. The direct utilisation of cyanide for isocyanide biosynthesis (Scheme 4, reaction e) then conversion to isothiocyanate 104, may also occur, but does not account for the significant labelling found in the thiocyanate 71. The interconversion of isocyanides and isothiocyanates by sponges may represent a mechanism for these organisms to adjust the concentrations of these metabolites for ecological purposes (see Section 4).
Advanced precursor studies in S. aurantium have been used to investigate the R–NC to R–NCS interconversion, and also the role proposed for isocyanides and isothiocyanates in dichloroimine biosynthesis in Scheme 5.96 Synthetic farnesyl amine (119)112 was used as a starting material for the radiochemical synthesis of formamide 115, isocyanide 120, and isothiocyanate 105. [14C]-Farnesyl isocyanide 120 was provided to S. aurantium and gave radioactive farnesyl isothiocyanate 105, in addition to the labelled stylotellanes 72 and 73. The absence of farnesyl isocyanide 120 as a natural product of S. aurantium, likely because of the instability of this compound, prevented assessment of the isothiocyanate→isocyanide transformation in this sponge. The incorporation of [14C]-farnesyl isothiocyanate 105 did, however, give radioactive stylotellane metabolites. The expected specific radiolabelling in the dichloroimine carbon was confirmed for the isolated samples of stylotellane B (72) by conversion (0.1 N H3PO4/MeOH) to the labelled cyclic carbamate 121 and the unlabelled amine 119. The labelling results are in accordance with Scheme 5 which details mechanisms for linear dichloroimine biosynthesis.90,96 The bicyclic metabolites 84 and 87 were also labelled in these advanced precursor experiments. Scheme 6 shows plausible mechanisms by which monocyclic and bicyclic dichloroimine metabolites might be generated from linear precursors.
 | ||
| Scheme 6 Biosynthetic scheme leading to cyclic dichloroimines. | ||
3.3 Biosynthesis in marine molluscs
As explored in Section 2, marine molluscs, notably nudibranchs, are also rich sources of terpene isocyanides and related metabolites. The isolation of the same metabolites from nudibranchs and sponges provides strong circumstantial evidence that the nudibranch isocyanide metabolites are of dietary origin.113,114 Experimental support for this proposal is provided by a transfer experiment involving the tropical sponge A. cavernosa and the nudibranch Phyllidiella pustulosa. The mollusc was allowed to feed for 5 days on sponges that had been previously incubated with [14C]-cyanide or [14C]-thiocyanate. Radiolabelled axisonitrile-3 (23) and axisothiocyanate-3 (24) were detected in the molluscs, consistent with their transfer from the sponge. In control experiments, injection of [14C]-cyanide or [14C]-thiocyanate into P. pustulosa did not yield radioactive metabolites. Thus, P. pustulosa does not carry out de novo biosynthesis of these metabolites from cyanide.89 The acquisition of dietary metabolites by nudibranchs, which must reflect the ecological importance of the sequestered metabolites, is explored further in Section 4.3.There is no evidence yet as to the origin of the guanidine group in the unusual polyketide metabolites, triophamine (122)115–117 and limaciamine (123),118 which have been isolated from the molluscs Triopha catalinae and Limacia clavigera, or in related sponge terpenoid guanidines such as the stellettadines 124–126.119,120 It would be of interest to determine whether cyanide/thiocyanate metabolism or amino acid metabolism is the source of the guanidine group in these metabolites.
A number of elegant stable isotope labelling studies by the Andersen, Faulkner and Cimino groups has confirmed that many species of nudibranch, mostly from temperate waters, are capable of de novo terpene biosynthesis. This topic has been comprehensively reviewed, however there are no reports of the de novo biosynthesis of terpene isocyanides.121–123
4 Chemical ecology of marine isocyanides and related compounds
The biological origin and roles of terpene isocyanides and related marine metabolites in the marine environment is of interest, given the biomedical activities frequently ascribed to these compounds. To date, these interesting metabolites have infrequently been candidates for rigorous ecological evaluation.4.1 Cellular location of metabolites
A question pertinent to both isocyanide biosynthesis and ecology is whether the bioactive metabolites are products of sponge or symbiont metabolism. The sponge Amphimedon terpenensis contains the cyanobacterial (= blue green algal) symbiont Aphanocapsa feldmanni in its outer tissue (where the photosynthetic activity of the cyanobacteria may contribute substantially to sponge nutrition) and high populations of diverse eubacteria in the inner tissue. When the various cell types present in the sponge were separated by gradient centrifugation on the artificial sugar Ficoll, diisocyanoadociane (54) was associated with the sponge cells present in the sponge; therefore the compound is likely biosynthesised by the sponge cells. It was speculated that the terpene might function as a structural component of sponge cell membranes.124 Electron microscopic studies show that Axinyssa n. sp. contains cyanobacteria in its surface tissue plus high bacterial populations in the core tissue while A. cavernosa and S. aurantium contain few bacterial symbionts. The different symbiont profiles of these biosynthetically-related sponges are entirely consistent with the terpenes being products of sponge rather than symbiont metabolism.4.2 Antifeedant, antifouling and toxicity studies
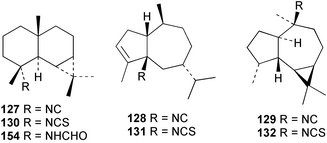
Faulkner et al. studied the chemical composition of the nudibranch Cadlina luteomarginata, and tested the feeding deterrency and toxicity of natural mixtures of three isocyanides (127–129) and of three isothiocyanates (130–132) isolated from the sponge diet of C. luteomarginata.125,126 The isocyanide mixture was an effective feeding deterrent against goldfish at 10 µg mL−1 and all compounds were toxic when tested at the high concentration of 100 µg mL−1. These laboratory assays suggested an ecological role for the compounds in situ. In an assay with the woolly sculpin Clinocottus analis, the isocyanide and isothiocyanate mixtures were partially effective as feeding deterrents.125 When the metabolites of the sponge A. cannabina and the nudibranch P. pulitzeri were tested in feeding inhibition and toxicity studies using the marine fish Chromis chromis and the freshwater fish Carassius carassius, the major sponge metabolite axisonitrile-1 (112) was ineffective as a feeding deterrent, but was toxic to fish.127 When extracts or metabolites of various phyllidid nudibranchs were assayed for ichthyotoxicity against the killifish Oryzias latipes, 9-isocyanopupukeanane (102) showed stronger activity then its 9-epi-isomer (133),128 while 2-isocyanoallopupukeanane (134) was toxic at 10 µg mL−1.10 Dichloromethane extracts of the Mediterranean sponge Acanthella acuta are toxic to the fish Lebistes reticulatus and kill dissociated cells of the freshwater sponge Ephydatia fluviatilis. The fish toxicity was traced to 1-isocyanoaromadendrane (135), which occurs in this sponge along with axisonitrile-3 (23).129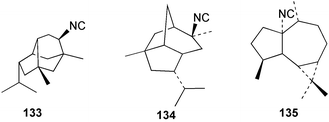
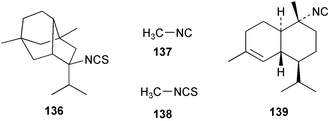
While extracts of the sponge Axinyssa sp. deterred feeding in the common pufferfish Canthigaster solandri, the major metabolite 5-isothiocyanatopupukeanane (136) did not deter feeding when tested at a relatively high concentration.130 A preliminary report of feeding deterrency tests on metabolites of A. cavernosa reveals that both the crude extract and sesquiterpene fraction were effective feeding deterrents, however axisonitrile-3 (23) alone was not an effective feeding deterrent; these data suggest a synergistic effect.89
Caribbean sponges of the genus Ircinia exude low molecular weight volatiles including methyl isocyanide (137) and methyl isothiocyanate (138) that give the sponge a characteristic odour, and which are responsible for the antimicrobial activity of the sponge extract. Exudation experiments carried out in situ and on sponges maintained under aquarium conditions revealed that these two compounds, and others, are continuously excreted by the sponge. When damaged, the concentration of these volatiles increases.131 While the ecological role of these volatiles in preventing fouling or infection has yet to be demonstrated,131 aquarium assays have shown that these compounds do not deter fish feeding.132 Paul and Rogers have commented that isocyanides are not effective feeding deterrents.133
Amine 11 from A. ambrosia, which is cytotoxic, has been shown to kill polyps of the scleractinian coral Madracis mirabilis.17 Exiguamide (26) from Geodia exigua inhibits cell fate specification during sea urchin embryogenesis at a minimum inhibitory concentration of 0.4 µM, but this activity was not shown by the related metabolites 27–29.29
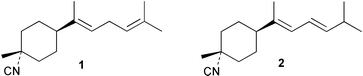
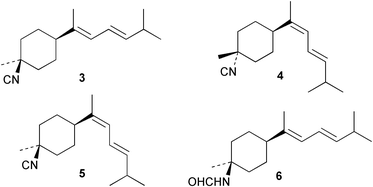
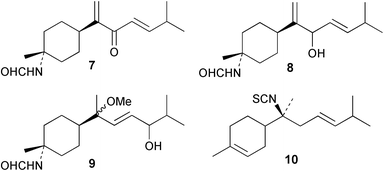
Laboratory assays involving the larvae of common invertebrates are frequently used to assess antifouling activity. A number of reports summarise the ability of metabolites of the sponge A. cavernosa to inhibit larval settlement and metamorphosis of the barnacle Balanus amphitrite.134,135 Sesquiterpenes such as 3-isocyanotheonellin (2) and 10-isocyano-4-cadinene (139), were antifouling at concentrations at which they were not toxic.11,12,135 Interestingly, structurally-related isothiocyanates were less active in these assays.135 The stereoisomers 3–5 of 3-isocyanotheonellin have also been evaluated against larvae of B. amphitrite and likewise showed high antifouling activity without significant toxicity.12 Axamide-3 (140) is reported to completely inhibit larval metamorphosis,40 while axisonitrile-3 (23) showed some activity. Other isocyanosesquiterpenes were less active.134
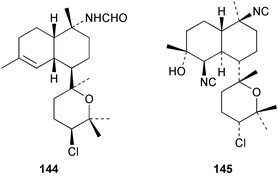
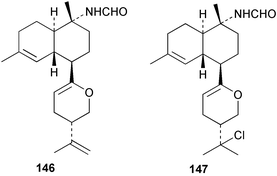
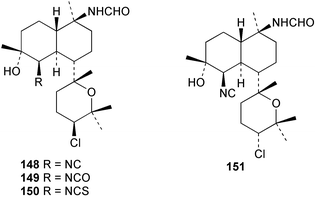
Among the diterpenes, metabolites of the kalihinol A and kalihinene series showed activity. Kalihinol A (30), 10-formamidokalihinene (141) and 15-formamidokalihinene (142) showed potent antifouling activity (LD50 ∼0.1 µg mL−1), were low in toxicity, and were more effective than copper(II) sulfate.134 The kalihinenes X (32), Y (143) and Z (144) were also active with EC50's of 0.49, 0.45 and 1.1 µg mL−1, as was kalihinol E (145). (0.088 µg mL−1).134,136,137 Kalihipyrans A (146) and B (147), both with formamido substituents, were active (IC50 1.3 and 0.85 µg mL−1 respectively), but less potent than isocyano-substituted metabolites.134,135,137 A number of other formamido-substituted kalihinols (148–151) also inhibited larval metamorphosis.40
Dichloroimine metabolites show useful antifouling activity. Fusetani et al. have reported that the axinyssamides A–C (74–76) showed fairly strong antifouling activity against B. amphitrite.138 Crude extracts from the dichloroimine-containing sponge S.aurantium have been shown to be inhibitors in a settlement assay using the ascidian Herdmania curvata,90 and reported to be of interest for their antifouling activity.67
The above examples demonstrate a range of potential benefits to sponges that produce bioactive nitrogenous terpene metabolites. It also seems likely that combinations of metabolites may be more effective than individual metabolites, but this has not yet been well tested. In particular, there may be ecological benefits to sponges that produce a range of metabolites, and which may have the ability to regulate the concentrations of these metabolites (cf.Axinyssa n.sp., as discussed in Section 3.2.2).
4.3 Sponge-nudibranch interactions
Johannes studied the toxic secretions of the nudibranch Phyllidia (=Phyllidiella) varicosa and found that the nudibranch was lethal to fish and crustaceans.139 Burreson et al. then observed P. varicosa feeding on the sponge Hymeniacidon sp., subsequently identified as Ciocalypta sp., and isolated 9-isocyanopupukeanane (102) from the sponge. Both the sponge and nudibranch contained isocyanides with a pronounced odour.140 Subsequently 2-isocyanopupukeanane (96) was also isolated from this sponge-nudibranch pair.141 Since then, many nudibranchs have been found to contain sponge-derived metabolites. A list of terpene isocyanide metabolites isolated from nudibranchs and their presumed sponge diets is shown in Table 1. The isolation of terpene isocyanides and isothiocyanates from both sponges and nudibranchs is circumstantial evidence that the nudibranchs acquire the metabolites by dietary transfer from sponges. The cyanide labelling experiment undertaken with Phyllidiella pustulosa, and described in Section 3.3, provides direct evidence for such a transfer process.89
| Nudibranch and metabolites | Probable sponge diet |
|---|---|
| Cadlina luteomarginata | |
| 127125,126 | Axinella sp. |
| 128125,126 | Axinella sp. |
| 129125,126 | Axinella sp. |
| Epipolasin-A (130)125,126 | Axinella sp. |
| 131125,126 | Axinella sp. |
| 132125,126 | Axinella sp. |
| 11-Isocyano-7β-H-eudesm-5-ene (17)143 | Acanthella sp. |
| 11-Isothiocyanato-7β-H-eudesm-5-ene (18)143 | Acanthella sp. |
| 127143 | Acanthella sp. |
| Acanthene C (152)143 | Acanthella sp. |
| 153143 | Acanthella sp. |
| 154143 | Acanthella sp. |
| Phyllidia sp. | |
| 3-Isocyanotheonellin (2)9 | Sponge not collected |
| Phyllidia bourguini | |
| 9-Isocyanopupukeanane (102)128 | Sponge not collected |
| 9-Epi-isocyanopupukeanane (133)128 | Sponge not collected |
| Phyllidiakrempfi | |
| 10-Isocyano-4-cadinene (139)11 | Sponge not collected |
| 10α-Isocyano-4-amorphene (155)11 | |
| Phyllidia ocellata | |
| 10α-Isocyano-4-amorphene (155)11 | Sponge not collected |
| 10α-Isocyano-4-amorphene (155)58 | Acanthella cf. cavernosa |
| Cavernothiocyanate (156)58 | Acanthella cf. cavernosa |
| Phyllidia pustulosa | |
| 3-Isocyanotheonellin (2)10,11 | Sponge not collected |
| 4α-Isocyanogorgon-11-ene (14)18 | Sponge not collected |
| 4α-Isothiocyanatogorgon-11-ene (15)18 | Sponge not collected |
| 4α-Formamidogorgon-11-ene (16)18 | Sponge not collected |
| 11-Isocyano-7β-H-eudesm-5-ene (17)18 | Sponge not collected |
| 11-Isothiocyanato-7β-H-eudesm-5-ene (18)18 | Sponge not collected |
| Axisonitrile-3 (23)11 | Sponge not collected |
| Axisothiocyanate (24)11 | Sponge not collected |
| 10-Epi-axisonitrile-3 (27)11 | Sponge not collected |
| 4-Thiocyanatoneopupukeanane (69)11 | Sponge not collected |
| 2-Thiocyanatoneopupukeanane (71)11 | Sponge not collected |
| 9-Isocyanopupukeanane (102)10 | Sponge not collected |
| 9-Epi-isocyanopupukeanane (133)10 | Sponge not collected |
| 2-Isocyanoallopupukeanane (134)10 | Sponge not collected |
| 10-Isocyano-4-cadinene (139)11 | Sponge not collected |
| 3-Isocyanobisabolane-8,10-diene (157)18 | Sponge not collected |
| 7-Isothiocyanato-7,8-dihydro-α-bisabolene (158)18 | Sponge not collected |
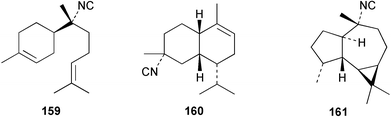
| 7-Isocyano-7,8-dihydro-α-bisabolene (159)10 | Sponge not collected |
| 4α-Isocyano-9-amorphene (160)10 | Sponge not collected |
| Axisonitrile-2 (161)10 | Sponge not collected |
| Axisonitrile-3 (23)89 | Acanthella cavernosa |
| Axisothiocyanate-3 (24)89 | Acanthella cavernosa |
| Phyllidiella pustulosa | |
| 9-Isocyanopupukeanane (102) and others51 | Axinyssa n.sp. |
| 10-Isothiocyanato-4-cadinene (22)22 | Phakellia carduus |
| Axisonitrile-3 (23)22 | Phakellia carduus |
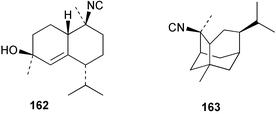
| 10-Isocyano-5-cadinen-4-ol (162)138 | Sponge not collected |
| Phyllidia pulitzeri | |
| Axisonitrile-1 (112)127 | Axinella cannabina4 |
| Phyllidia varicosa | |
| 4α-Isocyanogorgon-11-ene (14)18 | Sponge not collected |
| 4α-Formamidogorgon-11-ene (16)18 | Sponge not collected |
| 10-Isocyano-4-cadinene (139)11 | Sponge not collected |
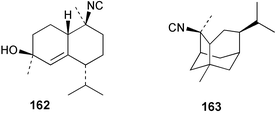
| 2-Isocyanotrachyopsane (163)11 | Sponge not collected |
| 2-Isocyanopupukeanane (96)141 | Hymeniacidon sp. (syn Ciocalypta) |
| 9-Isocyanopupukeanane (102)140,141 | Hymeniacidon sp. (syn Ciocalypta) |
| Reticulidia fungia | |
| 7965 | Sponge not collected |
| 8465 | Sponge not collected |
| Reticulidin A (87)65 | Sponge not collected |
| Reticulidin B (88)65 | Sponge not collected |
Sequestration of sponge compounds by nudibranchs such as P. varicosa and P. pustulosa potentially provides the molluscs with a feeding deterrent. Gunthorpe and Cameron studied the toxicity of Australian nudibranchs and found that Phyllidids were toxic to Gambusia affinus, but did not identify the chemical basis of the toxicity.142 Molluscs are known to selectively take up sponge metabolites,113 but the proposal that nudibranchs acquire dietary terpenes for chemical defence purposes has not yet been rigorously tested.133 Nudibranch metabolites need to be tested for alternative ecological roles; for example metabolites may perhaps act as chemical cues for feeding or larval settlement.113
4.4 Miscellaneous
In a useful paper that describes a closed-cycle aquaculture system for maintaining marine sponges, the ability of the sponge A. cavernosa to manufacture kalihinols when stressed by squeezing or damage was investigated. Other sponges were subjected to various light regimes. In these light and damage experiments the yields of kalihinol obtained decreased, compared to controls. A non-destructive method for “milking” these metabolites from living sponges was described.32 An earlier preliminary report has suggested chemical differences between wild type A. cavernosa and specimens maintained under the above controlled aquarium conditions for a seven month period.425 Acknowledgments
MJG thanks Professor Martin Banwell (Research School of Chemistry, ANU) for his hospitality during a sabbatical period, and acknowledges financial support from the Australian Research Council and The University of Queensland, and a Visiting Fellowship from the Research School of Chemistry, ANU. JSS thanks the Research School of Chemistry, ANU for funding.6 References
- C. W. J. Chang and P. J. Scheuer, in Topics in Current Chemistry, ed. P. J. Scheuer, Springer-Verlag, Berlin, 1993, p. 33 Search PubMed.
- C. W. J. Chang, in Prog. Chem. Org. Nat. Prod. ed. W. Herz, H. Falk, G. W. Kirby, and R. E. Moore, Springer, New York, 2000, p. 1 Search PubMed.
- M. J. Garson, J. S. Simpson, A. E. Flowers and E. J. Dumdei in Studies in Natural Products Chemistry, ed. Atta-ur-Rahman, Elsevier, Amsterdam, 2001, p. 329 Search PubMed.
- F. Cafieri, E. Fattorusso, S. Magno, C. Santocroce and D. Sica, Tetrahedron, 1973, 29, 4259 CrossRef CAS.
- D. J. Faulkner, Tetrahedron, 1977, 33, 1433 CrossRef CAS.
- M. S. Edenborough and R. B. Herbert, Nat. Prod. Rep., 1988, 5, 229 RSC.
- C. W. J. Chang and P. J. Scheuer, Comp. Biochem. Physiol., 1990, 97B, 227 Search PubMed.
- M. Iwashima, I. Terada, K. Iguchi and T. Yamori, Chem. Pharm. Bull., 2002, 50, 1286 CrossRef CAS.
- N. K. Gulavita, E. D. de Silva, M. R. Hagadone, P. Karuso, P. J. Scheuer, G. D. Van Duyne and J. Clardy, J. Org. Chem., 1986, 51, 5136 CrossRef CAS.
- N. Fusetani, H. J. Wolstenholme, S. Matsunaga and H. Hirota, Tetrahedron Lett., 1991, 32, 7291 CrossRef CAS.
- T. Okino, E. Yoshimura, H. Hirota and N. Fusetani, Tetrahedron, 1996, 52, 9447 CrossRef CAS.
- Y. Kitano, T. Ito, T. Suzuki, Y. Nogato, K. Shinsima, E. Yoshimura, K. Chiba, M. Tada and I. Sakaguchi, J. Chem. Soc., Perkin Trans. 1, 2002, 2251 RSC.
- H. Nakamura, J. Kobayashi, Y. Ohizumi and Y. Hirata, Tetrahedron Lett., 1984, 25, 5401 CrossRef CAS.
- C.-J. Li, F. J. Schmitz and M. Kelly, J. Nat. Prod., 1999, 62, 1330 CrossRef CAS.
- B. W. Sullivan, D. J. Faulkner, K. T. Okamoto, M. H. M. Chen and J. Clardy, J. Org. Chem., 1986, 51, 5134 CrossRef CAS.
- C. M. De Oliviera, C. C. Da Silva, C. H. Collins and A. J. Marsaioli, J. Braz. Chem. Soc., 2001, 12, 661 Search PubMed.
- N. V. Petrichtcheva, C. Duque, A. Dueñas, S. Zea, N. Hara and Y. Fujimoto, J. Nat. Prod., 2002, 65, 851 CrossRef CAS.
- K. E. Kassühlke, B. C. M. Potts and D. J. Faulkner, J. Org. Chem., 1991, 56, 3747 CrossRef CAS.
- P. Ciminiello, E. Fattorusso, S. Magno and L. Mayol, Can. J. Chem., 1987, 65, 518 CAS.
- X. Fu, J. R. Barnes, M. B. Hossain, F. J. Schmitz and D. Van der Helm, Nat. Prod. Lett., 1999, 12, 75 Search PubMed.
- R. J. Clark, B. L. Stapleton and M. J. Garson, Tetrahedron, 2000, 56, 3071 CrossRef CAS.
- A. D. Wright, Comp. Biochem. Physiol., 2003, 134A, 307 Search PubMed.
- D. Caine and H. Deutsch, J. Am. Chem. Soc., 1978, 100, 8030 CrossRef CAS.
- C. K. Angerhofer, J. M. Pezzuto, G. M. König, A. D. Wright and O. Sticher, J. Nat. Prod., 1992, 55, 1787 CrossRef CAS.
- G. M. König, A. D. Wright and S. G. Franzblau, Planta Med., 2000, 66, 337 CrossRef CAS.
- B. J. Burreson, C. Christopherson and P. J. Scheuer, J. Am. Chem. Soc., 1975, 97, 201 CrossRef CAS.
- J. Peng, X. Shen, K. A. El Sayed, D. C. Dunbar, T. L. Perry, S. P. Wilkins, M. T. Hamann, S. Bobzin, J. Huesing, R. Camp, M. Prinsen, D. Krupa and M. A. Wideman, J. Agric. Food Chem., 2003, 51, 2246 CrossRef CAS.
- K. A. El Sayed, D. C. Dunbar, D. K. Goins, C. R. Cordova, T. L. Perry, K. J. Wesson, S. C. Sanders, S. A. Janus and M. T. Hamann, J. Nat. Toxins, 1996, 5, 261 Search PubMed.
- M. M. Uy, S. Ohta, M. Yanai, E. Ohta, T. Hirata and S. Ikegami, Bioorg. Med. Chem. Lett., 2002, 12, 3037 CrossRef CAS.
- M. M. Uy, S. Ohta, M. Yanai, E. Ohta, T. Hirata and S. Ikegami, Tetrahedron, 2003, 59, 731 CrossRef CAS.
- S. Omar, C. Albert, T. Fanni and P. Crews, J. Org. Chem., 1988, 53, 5971 CrossRef CAS.
- D. Mendola, Biomol. Eng., 2003, 20, 441 Search PubMed.
- R. D. White and J. L. Wood, Org. Lett., 2001, 3, 1825 CrossRef CAS.
- M. Shimomura, H. Miyaoka and Y. Yamada, Tetrahedron Lett., 1999, 40, 8015 CrossRef CAS.
- C. W. Chang, A. Patra, J. A. Baker and P. J. Scheuer, J. Am. Chem. Soc., 1987, 109, 6119 CrossRef CAS.
- H. Miyaoka, H. Shida, N. Yamada, H. Mitome and Y. Yamada, Tetrahedron Lett., 2002, 43, 2227 CrossRef CAS.
- H. Miyaoka, M. Shimomura, H. Kimura and Y. Yamada, Tetrahedron, 1998, 54, 13467 CrossRef CAS.
- J.-C. Braekman, D. Daloze, F. Gregoire, S. Popov and R. Van Soest, Bull. Soc. Chim. Belg., 1994, 103, 187 CAS.
- M. L. Ciavatta, A. Fontana, R. Puliti, G. Scognamiglio and G. Cimino, Tetrahedron, 1999, 55, 12629 CrossRef CAS.
- H. Hirota, Y. Tomono and N. Fusetani, Tetrahedron, 1996, 52, 2359 CrossRef CAS.
- K. Kodama, R. Higuchi, T. Miyamoto and R. W. M. Van Soest, Org. Lett., 2002, 5, 169 CrossRef CAS.
- J. Rodriguez, R. M. Nieto, L. M. Hunter, M. C. Diaz, P. Crews, E. Lobkovsky and J. Clardy, Tetrahedron, 1994, 50, 11079 CrossRef CAS.
- S. J. Wratten, D. J. Faulkner, K. Hirotsu and J. Clardy, Tetrahedron Lett., 1978, 4345 CrossRef CAS.
- E. J. Corey and P. A. Magriotis, J. Am. Chem. Soc., 1987, 109, 287 CrossRef CAS.
- C. J. R. Fookes, M. J. Garson, J. K. MacLeod, B. W. Skelton and A. H. White, J. Chem. Soc., Perkin Trans. 1, 1988, 1003 RSC.
- L. Ciasullo, A. Cutignano, A. Casapullo, R. Puliti, C. A. Mattia, C. Debitus, R. Riccio and L. Gomez-Paloma, J. Nat. Prod., 2002, 65, 1210 CrossRef CAS.
- T. F. Molinski, D. J. Faulkner, G. D. Van Duyne and J. Clardy, J. Org. Chem., 1987, 52, 3334 CrossRef CAS.
- G. M. König, A. D. Wright and C. K. Angerhofer, J. Org. Chem., 1996, 61, 3259 CrossRef.
- H. A. Sharma, J. Tanaka, T. Higa, A. Lithgow, G. Bernardinelli and C. W. Jefford, Tetrahedron Lett., 1992, 33, 1593 CrossRef CAS.
- A. D. Wright, G. M. König, C. K. Angerhofer, P. Greenidge, A. Linden and R. Desqueyroux-Faúndez, J. Nat. Prod., 1996, 59, 710 CrossRef CAS.
- J. S. Simpson, M. J. Garson, J. N. A. Hooper, E. I. Cline and C. K. Angerhofer, Aust. J. Chem., 1997, 50, 1123 CAS.
- A. D. Wright, H. Wang, M. Gurrath, G. M. König, G. Kocak, G. Neumann, P. Loria, M. Foley and L. Tilley, J. Med. Chem., 2001, 44, 973 CrossRef.
- O. Schwarz, R. Brun, J. W. Bats and H.-G. Schmalz, Tetrahedron Lett., 2002, 43, 1009 CrossRef CAS.
- C. Singh, N. Chandra Srivastav and S. K. Puri, Bioorg. Med. Chem. Lett., 2002, 12, 2277 CrossRef CAS.
- H. He, D. J. Faulkner, J. S. Shumsky, K. Hong and J. Clardy, J. Org. Chem., 1989, 54, 2511 CrossRef CAS.
- H. He, J. Salvá, R. F. Catalos and D. J. Faulkner, J. Org. Chem., 1992, 57, 3191 CrossRef CAS.
- A. T. Pham, T. Ichiba, W. Y. Yoshida, P. J. Scheuer, T. Uchida, J. Tanaka and T. Higa, Tetrahedron Lett., 1991, 32, 4843 CrossRef CAS.
- N. Fusetani, H. J. Wolstenholme, K. Shinoda, N. Asai, S. Matsunaga, H. Onuki and H. Hirota, Tetrahedron Lett., 1992, 33, 6823 CrossRef CAS.
- A. Srikrishna and S. J. Gharpure, Tetrahedron Lett., 1999, 40, 1035 CrossRef CAS.
- A. Srikrishna and S. J. Gharpure, J. Chem. Soc., Perkin Trans. 1, 2000, 3191 RSC.
- S. J. Wratten and D. J. Faulkner, J. Am. Chem. Soc., 1977, 99, 7367 CrossRef CAS.
- S. Kehraus, G. M. König and A. D. Wright, J. Nat. Prod., 2001, 64, 939 CrossRef CAS.
- M. Musman, J. Tanaka and T. Higa, J. Nat. Prod., 2001, 64, 111 CrossRef CAS.
- A. Brust and M. J. Garson, ACGC Res. Commun., 2003, in press Search PubMed.
- J. Tanaka and T. Higa, J. Nat. Prod., 1999, 62, 1339 CrossRef CAS.
- W. Plansangkate, B. L. Stapleton and M. J. Garson, ACGC Res. Commun., 2001, 13, 23 Search PubMed.
- T. Higa, J. Tanaka, I. Ohtani, M. Musman, M. C. Roy and I. Kuroda, Pure Appl. Chem., 2001, 73, 589 Search PubMed.
- J. Tanaka and T. Higa, PCT/GB01/1213, 2001.
- M. Paîs, C. Fontaine, D. Laurent, S. La Barre and E. Guittet, Tetrahedron Lett., 1987, 28, 1409 CrossRef CAS.
- G. R. Pettit, J. K. Srirangam, D. L. Herald, K. L. Erickson, D. L. Doubek, J. M. Schmidt, L. P. Tackett and G. J. Bakus, J. Org. Chem., 1992, 57, 7217 CrossRef CAS.
- G. R. Pettit, J. K. Srirangam, D. L. Herald, K. L. Erickson, D. L. Doubek, J. M. Schmidt, L. P. Tackett and G. J. Bakus, J. Org. Chem., 1993, 58, 3222 CrossRef CAS.
- G. R. Pettit, J. K. Srirangam, D. L. Herald, J.-P. Xu, M. R. Boyd, Z. Cichacz, Y. Kamano, J. M. Schmidt and K. L. Erickson, J. Org. Chem., 1995, 60, 8257 CrossRef CAS.
- R. B. Kinnel, H.-P. Gehrken and P. J. Scheuer, J. Am. Chem. Soc., 1993, 115, 3376 CrossRef CAS.
- R. B. Kinnel, H.-P. Gehrken, R. Swali, G. Skoropowski and P. J. Scheuer, J. Org. Chem., 1998, 63, 3281 CrossRef CAS.
- T. Kato, Y. Shizuri, H. Izumida, A. Yokoyama and M. Endo, Tetrahedron Lett., 1995, 36, 2133 CrossRef CAS.
- D. H. Williams and D. J. Faulkner, Nat. Prod. Lett., 1996, 9, 57 Search PubMed.
- A. D. Patil, A. J. Freyer, L. Killmer, G. Hofmann and R. K. Johnson, Nat. Prod. Lett., 1997, 9, 201 Search PubMed.
- J. Tabrudravu, L. A. Morris, J. Kettenes van-den Bosch and M. Jaspars, Tetrahedron Lett., 2001, 42, 9273 CrossRef CAS.
- J. Tabrudravu, L. A. Morris, J. Kettenes van-den Bosch and M. Jaspars, Tetrahedron, 2002, 58, 7863 CrossRef.
- I. C. Piña, J. T. Gautschi, G.-Y.-S. Wang, M. L. Sanders, F. J. Schmitz, D. France, S. Cornell-Kennon, L. C. Sambucetti, S. W. Remiszewski, L. B. Perez, K. W. Bair and P. Crews, J. Org. Chem., 2003, 68, 3866 CAS.
- C. Li and A. J. Blackman, Aust. J. Chem., 1994, 47, 1355 CAS.
- C. Li and A. J. Blackman, Aust. J. Chem., 1995, 48, 955 CAS.
- H. Abe, S. Aoyagi and C. Kibayashi, J. Am. Chem. Soc., 2000, 122, 4583 CrossRef CAS.
- J.-L. Maeng and R. L. Funk, Org. Lett., 2002, 4, 331 CrossRef CAS.
- R. J. Capon, C. Skene, E. H.-T. Liu, E. Lacey, J. H. Gill, K. Heiland and T. Friedel, J. Org. Chem., 2001, 66, 7765 CrossRef CAS.
- P. Karuso and P. J. Scheuer, Tetrahedron Lett., 1987, 28, 4633 CrossRef CAS.
- M. R. Hagadone, P. J. Scheuer and A. Holm, J. Am. Chem. Soc., 1984, 106, 2447 CrossRef CAS.
- M. J. Garson, J. Chem. Soc., Chem. Commun., 1986, 35 RSC.
- E. J. Dumdei, A. E. Flowers, M. J. Garson and C. J. Moore, Comp. Biochem. Physiol., 1997, 118A, 1385 Search PubMed.
- J. S. Simpson, P. Raniga and M. J. Garson, Tetrahedron Lett., 1997, 38, 7947 CrossRef CAS.
- J. S. Simpson and M. J. Garson, Tetrahedron Lett., 1998, 39, 5819 CrossRef CAS.
- C. J. Silva, L. Wunsche and C. Djerassi, Comp. Biochem. Physiol., 1991, 99B, 763 Search PubMed.
- P. Karuso and P. J. Scheuer, J. Org. Chem., 1989, 54, 2092 CrossRef CAS.
- G. Cimino and G. Sodano, in Marine Natural Products – Diversity and Biosynthesis, ed. P. J. Scheuer, Springer-Verlag, Berlin, 1993, p. 77 Search PubMed.
- A. Iengo, C. Santacroce and G. Sodano, Experientia, 1979, 35, 10 Search PubMed.
- A. Brust and M. J. Garson, Tetrahedron Lett., 2003, 44, 327 CrossRef CAS.
- J. S. Simpson, PhD thesis, The University of Queensland, 2000.
- J. Westley, in Cyanide in Biology, ed. B. Vennesland, E. E. Conn, C. J. Knowles, J. Westley and J. Wissing, Academic Press, London, 1981, p. 61 Search PubMed.
- J. S. Simpson and M. J. Garson, Tetrahedron Lett., 1999, 40, 3909 CrossRef CAS.
- H. Ohkawa and J. E. Casida, Biochem. Pharmacol., 1971, 20, 1708 CrossRef CAS.
- J. R. Pollock and H. M. Goff, Biochem. Biophys. Acta, 1992, 1159, 279 Search PubMed.
- M. N. Hughes, in Chemistry and Biochemistry of Thiocyanic Acid and its Derivatives: ed. A. A. Newman, Academic Press, London, 1975, p. 2 Search PubMed.
- S. J. Wratten, D. J. Faulkner, D. Van Engen and J. Clardy, Tetrahedron Lett., 1978, 1391 CAS.
- E. Kühle, B. Anders and G. Zumach, Angew. Chem., Int. Ed., 1967, 6, 649 CrossRef.
- K.-H. van Pee and S. Unversucht, Chemosphere, 2003, 52, 299 CrossRef CAS.
- B. Vennesland, E. E. Conn, C. J. Knowles, J. Westley and J. Wissing, Cyanide in Biology, Academic Press, London, 1981 Search PubMed.
- J. P. Riley and G. Skirrow, Chemical oceanography, Academic Press, London, 1965 Search PubMed.
- V. Bornemann, G. M. L. Patterson and R. E. Moore, J. Am. Chem. Soc., 1988, 110, 2339 CrossRef CAS.
- M. J. Garson, V. Partali, S. Liaaen-Jensen and I. L. Stoilov, Comp. Biochem. Physiol., 1988, 91B, 293 Search PubMed.
- M. Rohmer, Nat. Prod. Rep., 1999, 16, 565 RSC.
- J. S. Simpson and M. J. Garson, Tetrahedron Lett., 2001, 42, 4267 CrossRef CAS.
- S. E. Sen and S. L. Roach, Synthesis, 1995, 756 CrossRef CAS.
- P. Karuso, in Bioorganic Marine Chemistry, ed. P. J. Scheuer, Springer-Verlag, Berlin, 1987, p. 31 Search PubMed.
- G. Cimino and G. Sodano, in Sponges in time and space, ed. R. W. M. Van Soest, T. M. G. Van Kempen and J. C. Braekman, Balkema, Amsterdam, 1994, p. 459 Search PubMed.
- K. Gustafson and R. J. Andersen, J. Org. Chem., 1982, 47, 2167 CrossRef CAS.
- J. Kubanek and R. J. Andersen, Tetrahedron Lett., 1997, 38, 6327 CrossRef CAS.
- E. I. Graziani and R. J. Andersen, J. Chem. Soc., Chem. Commun., 1996, 2377 RSC.
- E. I. Graziani and R. J. Andersen, J. Nat. Prod., 1998, 61, 285 CrossRef CAS.
- S. Tsukamoto, H. Kato, H. Hirota and N. Fusetani, Tetrahedron Lett., 1996, 37, 5555 CrossRef.
- S. Tsukamoto, T. Yamashita, S. Matsunaga and N. Fusetani, J. Org. Chem., 1999, 64, 3794 CrossRef CAS.
- G. Cimino, A. Fontana and M. Gavagnin, Curr. Org. Chem., 1999, 3, 327 Search PubMed.
- G. Cimino and M. Ghiselin, Chemoecology, 1999, 9, 187 CrossRef CAS.
- M. J. Garson, in Marine chemical ecology, ed. J. B. McClintock and B. J. Baker, CRC Press, Boca Raton, 2001, p. 71 Search PubMed.
- M. J. Garson, J. E. Thompson, R. M. Larsen, C. N. Battershill, P. T. Murphy and P. R. Berquist, Lipids, 1992, 27, 378 Search PubMed.
- J. E. Thompson, R. P. Walker, S. J. Wratten and D. J. Faulkner, Tetrahedron, 1982, 38, 1865 CrossRef CAS.
- D. J. Faulkner, T. F. Molinski, R. J. Andersen, E. J. Dumdei and E. D. de Silva, Comp. Biochem. Physiol., 1990, 97C, 233 Search PubMed.
- G. Cimino, S. De Rosa, S. De Stefano and G. Sodano, Comp. Biochem. Physiol., 1982, 73B, 471 Search PubMed.
- N. Fusetani, H. J. Wolstenholme and S. Matsunaga, Tetrahedron Lett., 1990, 31, 5623 CrossRef CAS.
- J.-C. Braekman, D. Daloze, F. Deneubourg, J. Huysecom and G. Vandevyver, Bull. Chim. Soc. Belg., 1987, 96, 539 Search PubMed.
- A. H. Marcus, T. F. Molinski, E. Fahy, D. J. Faulkner, C. Xu and J. Clardy, J. Org. Chem., 1989, 54, 5184 CrossRef CAS.
- C. Duque, A. Bonilla, E. Bautista and S. Zea, Biochem. Syst. Ecol., 2001, 29, 459 CrossRef CAS.
- J. R. Pawlik, G. McFall and S. Zea, J. Chem. Ecol., 2002, 28, 1103 Search PubMed.
- S. D. Rogers and V. J. Paul, Mar. Ecol. Prog. Ser., 1991, 77, 221 Search PubMed.
- N. Fusetani, H. Hirota, T. Okino, Y. Tomono and E. Yoshimura, J. Nat. Toxins, 1996, 5, 249 Search PubMed.
- N. Fusetani, Curr. Org. Chem., 1997, 1, 127 Search PubMed.
- T. Okino, E. Yoshimura, H. Hirota and N. Fusetani, Tetrahedron Lett., 1995, 36, 8637 CrossRef CAS.
- T. Okino, E. Yoshimura, H. Hirota and N. Fusetani, J. Nat. Prod., 1996, 59, 1081 CrossRef.
- H. Hirota, T. Okino, E. Yoshimura and N. Fusetani, Tetrahedron, 1998, 54, 13971 CrossRef CAS.
- R. E. Johannes, Veliger, 1963, 5, 104 Search PubMed.
- B. J. Burreson, P. J. Scheuer, J. Finer and J. Clardy, J. Am. Chem. Soc., 1975, 97, 4763 CrossRef CAS.
- M. R. Hagadone, B. J. Burreson, P. J. Scheuer, J. S. Finer and J. Clardy, Helv. Chim. Acta, 1979, 62, 2484 CrossRef.
- L. Gunthorpe and A. M. Cameron, Mar. Biol., 1987, 94, 39 CAS.
- D. L. Burgoyne, E. J. Dumdei and R. J. Andersen, Tetrahedron, 1993, 49, 4503 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |