Merging the potential of microbial genetics with biological and chemical diversity: an even brighter future for marine natural product drug discovery
Christine E. Salomona, Nathan A. Magarveya and David H. Sherman*b
aDepartment of Microbiology, University of Minnesota, Minneapolis, MN 55455, USA. E-mail: csalomon@umn.edu; magar001@umn.edu
bDepartment of Medicinal Chemistry, University of Michigan, Ann Arbor, MI 48109, USA. E-mail: davidhs@umich.edu
First published on 15th December 2003
Abstract
Covering: 1999–2003
Marine invertebrates and a growing number of marine bacteria are the sources of novel, bioactive secondary metabolites. Structurally, many of these compounds appear to be biosynthesized by polyketide synthases (PKS) and/or nonribosomal peptide synthetases (NRPS) that have also been found in terrestrial microbes. This review highlights scientific advances from 1999–2003 in the emerging field of molecular genetics of polyketide and nonribosomal peptide natural products isolated from marine organisms. The implications of this research towards the development of marine secondary metabolites as a sustainable source of new drugs are discussed.
 Christine E. Salomon | Christine E. Salomon, born in 1972 in Morristown, NJ, received her B.S. with honors in Marine Biology from Southampton College, Long Island University in 1995. After an undergraduate internship in the laboratory of the late Professor D. John Faulkner at Scripps Institution of Oceanography, University of California, she joined his research group to study the cellular localization of natural products in marine invertebrates and received her PhD in 2001. She is currently a MinnCResT NIH postdoctoral fellow in the laboratory of Professor David H. Sherman at the University of Minnesota. Her research interests include the molecular genetics of natural products, the location of their biosynthetic genes in complex microbial/invertebrate assemblages and their roles in the ecology and biology of marine organisms. |
 Nathan A. Magarvey | Nathan A. Magarvey was born in Annapolis Royal, Nova Scotia (Canada) in 1974. He studied biochemistry at Dalhousie University (Canada), working as a research assistant under Professor Leo C. Vining, studying the shikimic acid pathway derived antibiotic, chloramphenicol. Following this he moved to Pearl River, NY (USA) to work in the Natural Products Research section at Wyeth Research, studying the genetics and biochemistry of the lipoglycopeptide antibiotic, manno-peptimycin. In September 2001 he moved to the University of Minnesota (USA) for PhD studies under Professor David H. Sherman. In D. H. Sherman's laboratory, his research focuses on the chemistry and biology of marine actinomycetes and the biosynthesis of the cyanobacterial polyketide/peptide tubulin-binding agent, cryptophycin. |
 David H. Sherman | David H. Sherman obtained his undergraduate degree in Chemistry at the University of California, Santa Cruz, and completed his PhD in the Department of Chemistry at Columbia University with Professor Gilbert Stork. After postdoctoral studies at the Yale University School of Medicine and MIT, Dr Sherman spent three years at Biogen Research Corporation, and in 1987 moved to the John Innes Institute, Norwich, England to work with Professor Sir David Hopwood in microbial genetics. He began his academic career at the University of Minnesota in 1990, and in 2003 moved to the University of Michigan, Ann Arbor where he is the John G. Searle Jr. Professor of Medicinal Chemistry. |
1 Introduction
The genomics revolution has forever changed the face of terrestrial natural products research. In only the last two decades, the complete biosynthetic gene clusters of more than 150 secondary metabolites from bacteria, fungi and plants have been identified.1,2 Recent investigations (past 15 years) into the genetics of secondary metabolism have resulted in a series of new genetic tools and provided the stimulus for a broad resurgence in the field of natural product biosynthesis. Such molecular approaches have offered insight and new perspectives into old questions previously unanswered by “classical” precursor-driven feeding studies. In other cases, a complementary combination of the two techniques has served as a basis to re-employ chemical probing strategies in a more directed manner to understand not only biosynthetic strategies, but also timing of chemical transformations (i.e. precursor feeding studies in engineered modular polyketide systems).3,4 This merging of biological and chemical approaches has been developed almost exclusively in terrestrial microbial systems and is now being used to understand the biosynthesis of completely novel, complex metabolites isolated from marine organisms.5–7This molecular perspective has focused on some of the most pharmaceutically useful and structurally interesting terrestrial microbial metabolites belonging to the biosynthetic classes of polyketide synthases (PKSs) and nonribosomal peptide synthetases (NRPSs). Many of the complex, bioactive compounds isolated from marine organisms appear to arise from similar biosynthetic pathways, and may be approached using our knowledge of analogous terrestrial enzyme systems. Classical feeding studies using isotope labeled precursors continue to be a useful tool to elucidate the mechanisms of biosynthesis of marine metabolites from cultured organisms, but there is a growing trend towards the use of molecular genetics to identify biosynthetic pathways and novel enzymes. The ability to clone and heterologously express gene clusters encoding valuable metabolites may help to alleviate the critical supply problems associated with most marine natural products and provide contributions towards the “molecular toolbox” used in the genetic engineering of new drugs.
Currently, there are only three examples of published gene clusters encoding PKS and NRPS natural products from marine organisms, and these are exclusively from cultured marine bacteria.5–7 However, recent research suggests that the adaptation of metagenomic analysis, microbial genetic and culturing techniques to the unique systems in the marine environment will allow this number to increase dramatically in the coming years.8–10
Here we review some of the molecular approaches taken to understand the biosynthesis of metabolites encoded by PKS and NRPS gene clusters in marine organisms and present strategies for the discovery of additional pathways. Particular emphasis will focus on the structural parallels of terrestrial bacterial secondary metabolites and certain classes and types of marine natural products. The molecular underpinnings of these relationships will be addressed in two major areas: 1) natural product assembly line levels (“terrestrial” versus “marine” natural product biosynthesis), and 2) phylogenetic conservation of biosynthetic pathways. Stemming from this analysis we will discuss what role marine microbiology and bacterial genetics will play in the future of marine natural product drug discovery. In particular, how the power of these approaches will aid in realizing the potential of marine systems as a rich, tractable and sustainable source for drug discovery.
2 Marine natural products
Since the 1970's, more than 15,000 structurally diverse natural products with an astounding array of bioactivities have been discovered from marine microbes, algae and invertebrates.11 Unfortunately, the utility of marine natural products as a potentially sustainable drug source is hampered by several significant limitations. Compounds are often isolated in extremely low yields and may be difficult to synthesize economically. Moreover, source organisms (especially invertebrates and/or their symbionts) can be difficult to culture or may not produce the compound of interest under the given culture conditions, and wild collection of macroorganisms can be detrimental to the environment.12,13 Despite these disadvantages, interest in the development of marine compounds as potential drugs is thriving and there are several dozen marine natural products (or derivatives thereof) that are in clinical or preclinical trials for the treatment of cancer, inflammation and other diseases (Table 1).14,15 Didemnin B, isolated from a tunicate over 20 years ago, was the first marine natural product to enter human clinical trials against cancer and led the way for a plethora of drug candidates isolated from marine organisms.16| Compound | Probable biosynthetic route or compound type | Source organism | Disease target | Clinical phase status |
|---|---|---|---|---|
| Ziconotide (Prialt™) | Ribosomally produced peptide | Conus magnus (mollusc) | Neuropathic pain | III |
| Ecteinascidin 743 (Yondelis™) | NRPS | Ecteinascidia turbinata (tunicate) | Cancer | II/III |
| Bryostatin 1 | Mixed PKS/NRPS | Bugula neritina (bryozoan) | Cancer | II |
| Dolastatin 10 | NRPS | Dolabella auricularia (mollusc, and later found in cyanobacteria) | Cancer | II |
| Dehydrodidemnin B (Aplidine™) | NRPS | Aplidium albicans (tunicate) | Cancer | II |
| Kahalalide F | NRPS | Elysia rufescens/Bryopsis sp. (mollusc/green alga) | Cancer | II |
| Squalamine | aminosteroid | Squalus acanthias (shark) | Cancer | II |
| IPL-576092 (HRM-4011A) | steroid | Petrosia contignata (sponge) | Psoriasis | II |
| IPL-512602 | steroid | Petrosia contignata (sponge) | Asthma/inflammation | II |
| IPL-550260 | steroid | Petrosia contignata (sponge) | Inflammation | I |
| Halichondrin B derivative (synthetic, E7389) | PKS | Lissodendoryx sp. (sponge) | Cancer | I |
| Discodermolide | PKS | Discodermia dissoluta (sponge) | Cancer | I |
| Hemiasterlin derivative (HTI-286) | NRPS | Cymbastella sp. (sponge) | Cancer | I |
| Bengamide derivative (LAF389) | Mixed PKS/NRPS | Jaspis sp. (sponge) | Cancer | I |
| Agelasphin derivative (KRN-7000) | glycosphingolipid | Agelas mauritianus (sponge) | Cancer | I |
| Laulimalide | PKS | Cacospongia mycofijiensis (sponge) | Cancer | Preclinical |
| Vitilevuamide | NRPS | Didemnum cucliferum and Polysyncraton lithrostrotum (tunicates) | Cancer | Preclinical |
| Diazonamide | NRPS | Diazona angulata (tunicate) | Cancer | Preclinical |
| Eleutherobin | diterpene glycoside | Eleutherobia sp. and Erythropodium caribaeorum (soft corals) | Cancer | Preclinical |
| Sarcodictyin | terpene | Sarcodictyon roseum (sponge) | Cancer | Preclinical |
| Peloruside A | PKS | Mycale hentscheli (sponge) | Cancer | Preclinical |
| Salicylhalimides A and B | PKS | Haliclona sp. (sponge) | Cancer | Preclinical |
| ES-285 (Spisulosine) | alkyl amino alcohol | Spisula polynyma (mollusc) | Cancer | Preclinical |
| Thiocoraline | NRPS | Micromonospora marina (bacteria) | Cancer | Preclinical |
The supply of marine metabolites being tested in the clinic is currently provided by several means: open aquaculture of the invertebrates (ET-743 and bryostatin), total synthesis (ziconotide, discodermolide, dolastatin 10, dehydrodidemnin, hemiasterlins), semi-synthesis (halichondrin B derivative, ET-743), and fermentation of producing microbes (thiocoraline).17 In general, fermentation has been the most viable method for production of natural products (penicillin, clavulanic acid, erythromycin, etc.), especially if the compounds proceed through clinical trials and are needed on a commercial scale (> kg).18 Analysis of the structural types present in Table 1 suggests that more than half are polyketides and/or peptide metabolites, with predicted origins stemming from modular PKSs and NRPSs. Our rapidly growing knowledge of modular biosynthetic systems may allow these gene clusters to be cloned and overexpressed in more amenable bacterial hosts. This approach could potentially provide a virtually unlimited supply of compounds and alleviate the need for culturing of the source macroorganisms.
Although many marine natural products appear to arise from multi-functional enzymes that are also present in terrestrial systems, many also possess a myriad of functional groups not previously described from terrestrial metabolites. These unique structural differences are inevitably reflected in novel biosynthetic pathways with genes encoding completely new enzymatic activities.6,7,19 Despite these differences, the presence of conserved regions of “terrestrial” biosynthetic genes can be used for probing analogous pathways in marine systems both in vivo through molecular cloning and in silico by analyzing sequenced genes and genomes for secondary metabolic enzymes and pathways.
3 Co-linear assembly of natural products by PKS and NRPS
Currently, our understanding of the biosynthesis of complex natural products at both a genetic and chemical level is centered on those produced by modular type I PKS and NRPS systems. While the products of these pathways are very different in structure and function, they have in common a similar co-linear assembly using a “swing-arm” co-factor (phosphopantethiene) to enable a variety of the enzymatic activities to occur in a highly ordered manner on the surface of large, multifunctional proteins. These enzymes are organized in a modular fashion, and use specific domains to sequentially catalyze the condensation of simple carboxylic acids (PKS) or amino acid (NRPS) building blocks into a growing chain in an assembly-line manner. Each module in a cluster is responsible for chain extension through the recognition, activation and incorporation of specific substrates. Structural diversity is introduced by varying the combinations and permutations of both integral and auxiliary domains. Here we present a brief overview of both biosynthetic systems, but for greater detail the reader is directed to several excellent in-depth reviews (PKS20–22 and NRPS23–25).3.1 Polyketide biosynthesis
One unifying theme in the diverse polyketide family of metabolites is their biosynthesis through the sequential condensation of small carboxylic acids, reminiscent of the synthesis of fatty acids in bacteria, lower and higher eukaryotes.26 There are three major classes of PKS systems, arranged by their mode of synthesis and structural type of product. Type I PKSs in bacteria are multienzyme complexes that are organized into individual, linear modules, each of which is responsible for a single, specific chain elongation process and post condensation modification of the resulting β-carbonyl. The fungal type I PKSs consist of a single, giant protein that iteratively uses the same set of domains in a module for building the polyketide. Type II, or aromatic, PKSs are complexes of monofunctional proteins characterized by their iterative use of a single set of distinct enzymes to construct polyketide chains which are then cyclized to produce small molecules containing aromatic ring systems. Recently, a third type of polyketide synthase was identified from bacteria belonging to the chalcone and stilbene synthase family of enzymes from plants. These enzyme systems, unlike the Type I and II systems, contain one protein, one domain and one active site to carry out the three central reactions of chain initiation, elongation and cyclization.27The actions of the essential “core” domains present in types I and II PKS systems are presented schematically in Fig. 1. The initiation module begins with the actions of the acyl transferase domain (AT) selection of an activated acyl-CoA monomer (usually malonyl or methyl malonyl CoA). Following this selection step, the AT domain transfers the acyl-CoA to the adjacent “swinging arm” of the post-translationally (phosphopantethiene) modified acyl carrier protein (ACP). The chain is transferred from the ACP to the upstream ketosynthase domain (KS) required for catalyzing the decarboxylation of the carboxylic acid and subsequent Claisen condensation between the growing chain (or starter unit) and the downstream ACP tethered extender unit.
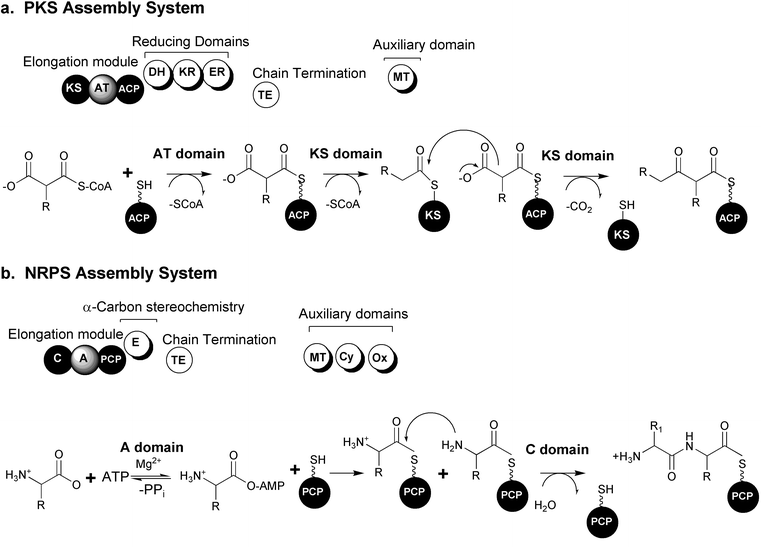 | ||
| Fig. 1 Modular biosynthetic pathway systems. (a) Polyketide synthase (PKS) organization and mechanisms. (b) Nonribosomal peptide synthetase (NRPS) organization and mechanisms. (KS: ketosynthase, AT: acyl transferase, ACP: acyl carrier protein, DH: dehydratase, KR: ketoreductase, ER: enoyl reductase, TE: thioesterase, MT: methyl transferase, C: condensation, A: adenylation, PCP: peptidyl carrier protein, E: epimerase, Cy: cyclization, Ox: oxidoreductase) (Figure adapted from Schwarzer et al., 2003).23 | ||
In type I PKSs, structural diversity is introduced in part by optional reductive domains acting upon the newly formed β-carbonyl after each extension reaction. The “reductive loop” can consist of one or more of the following activities: ketoreductase (KR) reduces the carbonyl to a hydroxyl group, a dehydratase (DH) dehydrates the alcohol to form a double bond and an enoyl reductase (ER) is responsible for the reduction of the double bond to a fully saturated methylene. The incorporation of unusual starter molecules (amino acids, fatty acid derivatives, etc.) and extension units also plays an important part in generating immense diversity in polyketide structures.28 Additional “auxiliary” domains consist of methyl transferases (MT) or other embedded activities.29 The complete elongated and functionalized chain is often transferred to a final thioesterase domain (TE) capable of catalyzing the hydrolytic release of a linear compound or a hydrolytic release coupled with cyclization to generate a macrolactone structure.
3.2 Nonribosomal peptide biosynthesis
Nonribosomally produced peptide metabolites display a remarkable spectrum of activities and are extremely important as pharmaceuticals.30–33 Examples of some of the most clinically useful peptides are the antibiotics vancomycin and penicillin, the immunosuppressive agent cyclosporine and the antitumor compound bleomycin. Compounds synthesized by NRPSs can be distinguished by the presence of non-proteinogenic, branched and D-amino acids and are often cyclic in structure.Like the Type I PKS systems, NRPSs are large, multifunctional enzyme complexes that build growing chains from individually selected building blocks. The NRPS is also organized into modules, each of which is responsible for one cycle of elongation by the incorporation of a single amino acid into the growing peptide chain. Each elongation module contains three essential domains: adenylation, thiolation and condensation. The adenylation domain (A) selects a specific amino acid and activates it as an amino acyl adenylate. The activated amino acid is then transferred to the phosphopantethiene group (“swing arm”) of the post-translationally modified peptidyl carrier protein (PCP) or thiolation domain (T). The condensation domain (C) catalyzes the peptide bond formation between amino acids in adjacent modules. The chain is elongated successively and released at the end of the “assembly-line” by the action of an integrated thioesterase (TE) domain or a separate TE generating either a linear or cyclic peptide. Additional structural diversity is introduced by modification of the growing chain catalyzed by a variety of embedded auxiliary domains such as epimerization (E), N-methylation (MT), cyclization (Cy), oxidoreductase (Ox), N-formylation (F) and reductase (R) domains.33 These domains provide further methods to deviate from the nonribosomal code, providing the incorporation of diverse amino acid functionality such as thiazoles, oxazoles, thiazolidines, oxazolidones, as well as a full array of N-methylated and D-amino acids not found in any other system throughout nature.33
4 Marine microbial gene clusters
For many years it was believed that only a very small percentage of microbes unique to the marine environment were culturable and it was not until a variety of marine-specific techniques were employed that a much greater number of diverse microbes could be isolated from seawater, marine sediments and invertebrates.34–37 These efforts have provided an increasing number of unique bioactive natural products from marine bacteria which are particularly attractive due to the availability of cultured source organisms.38 Many marine microbial metabolites possess both potent bioactivity and interesting polyketide and/or peptide structures and are therefore attractive targets for molecular genetic studies.The three complete gene clusters (PKS and NRPS) that have been reported from marine organisms were isolated from cultured actinomycetes and cyanobacteria, two microbial groups that are especially well known for their ability to produce diverse natural products. All of the clusters were identified from cosmid libraries by developing probes designed from conserved regions of biosynthetic domains from terrestrial microbial pathways. Although the quantity of published marine gene clusters is relatively small, we expect that there will be more examples in the near future. Work in our lab, in collaboration with William Gerwick's lab (Oregon State University), is currently involved with the identification of several new marine clusters for the microbial metabolites curacin A, carmabin and barbamide. In addition to complete clusters, there is an increasing number of examples of individual biosynthetic genes and putative sections of clusters that have been identified in several organisms, including dinoflagellates and a potential microbial symbiont of an invertebrate.9,10
The following marine bacterial gene clusters demonstrate that while PKS and NRPS biosynthetic routes are conserved between terrestrial and marine systems, in some cases there are additional, novel catalytic enzymes responsible for the unique functional groups found solely in marine natural products.6,7,39
4.1 Marine actinomycetes
Terrestrial actinomycetes have provided the highest number and diversity of unique, bioactive secondary metabolites and are the source of some of the most clinically useful pharmaceuticals. Recent investigations focusing on marine derived actinomycete isolates have yielded many new biologically active compounds, and more importantly, a surprisingly much improved rate of discovery of novel compounds versus their terrestrial counterparts.40
The enc cluster consists of 20 ORFs spanning 21.3 kb and was overexpressed in Streptomyceslividans. The core structure of enterocin is synthesized by an uncommon benzoate starter unit and extended by 7 malonate molecules (Fig. 2). At least five ORFs present in the enc cluster are involved in the synthesis (EncP,H,I,J) and incorporation (EncL) of the benzoic acid unit which is derived from L-phenylalanine via a plant-like oxidative pathway.46,47 The biosynthesis of the unusual benzoyl-CoA starter unit is reviewed by Moore and Hertweck in an earlier issue of this journal and illustrated in Fig. 2.28 An expected dehydrogenase for oxidation of the β-keto group on β-ketophenylpropionyl-CoA was not found and cyclases which are typical of all other type II PKS systems were also absent.
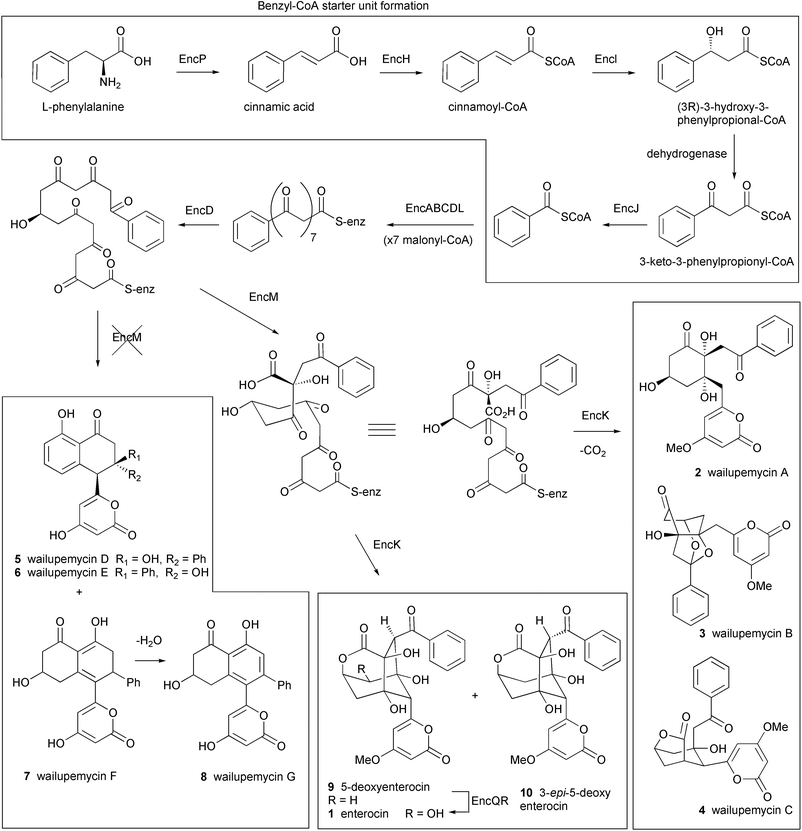 | ||
| Fig. 2 Proposed biosynthetic pathway for enterocin and the benzoyl-CoA starter unit. EncP: phenylalanine ammonia-lyase, EncH: cinnamate-CoA ligase, EncI: cinnamoyl-CoA hydratase, EncJ: 3-keto-3-phenylpropionyl-CoA thiolase, EncABCD: minimal PKS, EncL: aromatic acyl transferase, EncM: oxygenase, EncK: methyl transferase, EncQ: ferredoxin, EncR: cytochrome P-450 hydroxylase. | ||
Earlier precursor feeding studies by Seto et al. suggested that enterocin is derived from a linear precursor which undergoes an unusual Favorskii-like carbon rearrangement.48 This rare carbon rearrangement reaction is proposed to be involved in the biosynthesis of several natural products, including the toxin okadaic acid isolated from dinoflagellates.49 It is presumed that such a rearrangement prevents the successive aldol condensations of reactive intermediates that typically lead to an aromatic structure in all other type II PKS systems. The identity of the putative “favorskiiase”, EncM, was shown by gene knock out studies and the successive isolation of wailupemycin D 5 (previously isolated from a natural strain) and novel non-rearranged analogues wailupemycins E–G 6–8.42 The encoded methyltransferase, EncK, was proposed to be responsible for O-methylation of the α-pyrone ring and is possibly linked to the EncM mediated rearrangement leading eventually to enterocin, 5-deoxyenterocin 9 and 3-epi-5-deoxyenterocin 10.19 The enterocin cluster is the first example of a “derailed” aromatic polyketide synthase which encodes natural chemical diversity by the versatility of associated tailoring enzymes and pathway branch points.
The 34 kb grh type II PKS cluster was localized to a single cosmid and contained 33 ORFs. The loading molecule is an acetate which is elongated by 12 malonyl-CoA molecules via the core PKS genes (grhABC) (Fig. 3). The ACP gene grhC is unusual due to its location 26 kb upstream of the ketosynthase domains grhA and grhB. The extended chain is then putatively cyclized by products of two different aromatase genes, grhE and grhQ, and the associated genes grhSTL to yield a pradimicin-like precursor. An unprecedented number of genes encoding a variety of oxidoreductases (11) were identified as responsible for the many oxidative steps required to produce the epoxyspiroketal and naphthoquinone ring systems. The authors hypothesize that one possible oxidative degradation pathway may involve a retro aldol reaction before two Baeyer–Villiger oxygen insertions leading to the excision of a C2 unit and subsequent cyclization to form the spiroketal. A putative P450 monooxygenase is suggested to catalyze the epoxidation of the C19 double bond. The identification of the cluster was confirmed by overexpression of the cosmid clone in S. lividans and production of griseorhodin A 11.
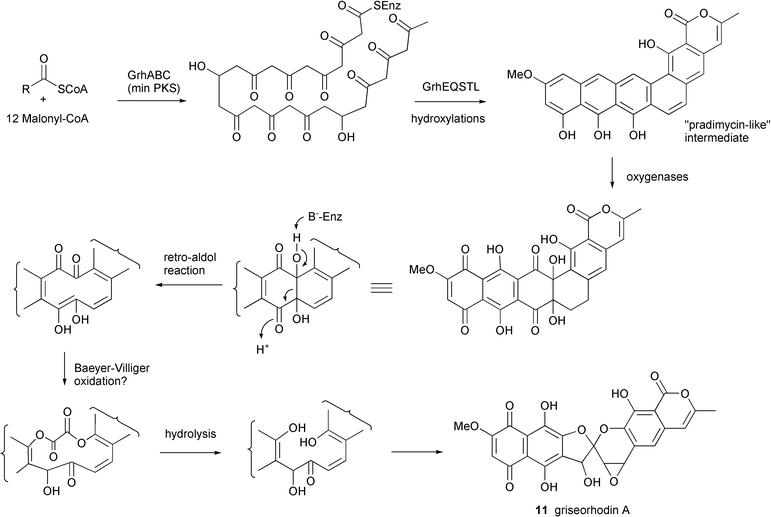 | ||
| Fig. 3 Proposed biosynthetic pathway for griseorhodin A. | ||
4.2 Marine cyanobacterial pathways
Terrestrial cyanobacteria are well known for their ability to produce cytotoxic nonribosomally derived peptides and depsipeptides.53,54 Analysis of the published genome of the cyanobacterium Nostocpunctiforme suggests that there are at least 16 distinct modular NRPS/PKS clusters present in this single organism.55 Cyanobacteria from the marine environment are also prolific sources of unique, biologically active peptide and polyketide metabolites and have yielded more than 450 novel metabolites.11,38 The ability to culture several species of marine cyanobacteria, especially members of the metabolite-rich genus Lyngbya, has facilitated both molecular genetic studies and labeled precursor biosynthetic experiments.
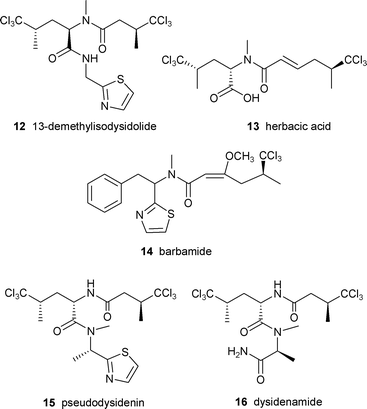
In order to gain understanding of the origin of the unusual trichloro-amino acid and biosynthesis of this group of compounds, Sitachitta and coworkers conducted several biosynthetic studies with a Curaçao strain of L. majuscula that produced the lipopeptide barbamide 14 in culture.61 Stable isotope labeled precursor feeding experiments established that the trichloromethyl moiety was derived from the pro-R methyl group of L-leucine.61 Similar experiments with labeled [2-13C] 5,5,5-trichloroleucine demonstrated high incorporation rates, suggesting its role as a direct intermediate. Other feeding studies showed the additional incorporation of L-cysteine, L-phenylalanine, acetate and S-adenosylmethionine, signifying a mixed PKS/NRPS biosynthetic origin.61
An interest in the unusual biosynthesis and bioactivity of barbamide led Sherman and coworkers to identify and clone the barbamide gene cluster.7 One of the challenges associated with the cluster was the variety of compounds that were produced by the same strain and were presumably also the products of mixed PKS/NRPS systems including curacin A, carmabin, antillatoxin and malyngamide. The number of clones positive to both the PKS and NRPS probes was narrowed down by focusing on adenylation sequences specific for cysteine, presumed to be involved in thiazole ring formation.
The bar gene cluster was found to span 40 kb and was isolated as two cosmid clones. Twelve ORFs, designated barA–barH, were identified as the putative core genes involved in barbamide production (Fig. 4). Leucine is presumably selected and activated by the BarD adenylation domain and loaded onto the BarA PCP. The leucine is then possibly chlorinated by the actions of BarB1, BarB2 and BarC and transferred to the NRPS/PKS hybrid loading module, BarE. While the trichloroleucine is tethered to the PCP of BarE, BarJ may oxidatively deaminate the amino acid. A rare one-carbon truncation is postulated to then occur by decarboxylation of the α-keto intermediate. The resulting trichloroisovaleryl group may be extended by a malonyl unit forming a diketide. An O-methylation domain in BarF could subsequently catalyze formation of the enol methyl ether to stabilize the enol form of the β-carbonyl. The role of BarF in the formation of the energetically unfavorable C4–C5 E double bond is not yet understood. The chain is further extended by phenylalanine which is presumably N-methylated by the actions of the NRPS module BarG and then condensed with a final cysteine residue which is cyclized to form the thiazoline ring. According to this putative biosynthetic plan, the molecule must undergo a final decarboxylation to yield barbamide, which may be catalyzed by a potential oxidative domain (BarJ). In the absence of gene disruption or heterologous expression, the identification of the cluster was supported by expression of the NRPS adenylation domains and subsequent assay for ATP-[32P]PPi exchange activity. The amino acid specificity of each A domain (leucine, trichloroleucine and cysteine) correlated with the putative biosynthetic assembly of barbamide.
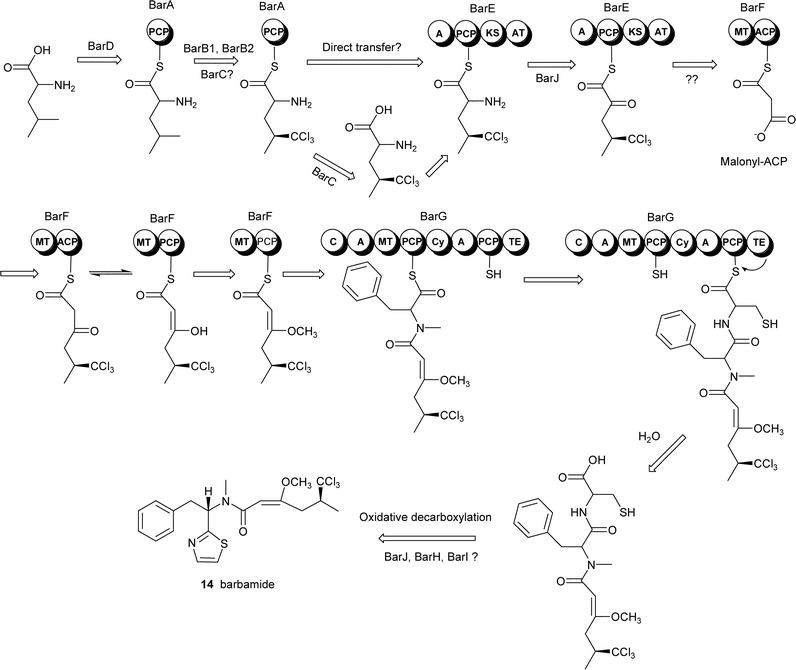 | ||
| Fig. 4 Proposed biosynthetic pathway for the cyanobacterial lipopeptide barbamide. Reprinted from Chang et al., 2002 with permission from Elsevier.7 | ||
The gene clusters shown above clearly reveal that at least some marine bacteria utilize the same modular biosynthetic pathways that are used by terrestrial bacteria. This realization demonstrates that the growing number of novel, cultured marine bacteria may provide a completely new, tractable resource for molecular genetic studies. Additionally, there is tantalizing evidence that many compounds from marine invertebrates may also be produced by bacteria (see below): such pathways could potentially be obtained using similar molecular techniques to harness an even greater wealth of chemical and enzyme diversity.
5 Determination of biosynthetic sources of natural products in marine invertebrates
Are the secondary metabolites isolated from marine invertebrates produced by microbes? This is a long standing question among marine natural products chemists and has often been proposed for metabolites isolated from sponges, ascidians, soft corals and bryozoans.10,62,63 Many compounds from marine macro-organisms bear structural resemblance to those from terrestrial (and marine) microbes.62–65 These observations invariably invoke the hypothesis of a microbial symbiont as the biosynthetic source of the compound identified from the invertebrate. The circumstantial evidence for microbial origins of these compounds abounds, though examples of direct confirmation are rare.66 Here we present additional support by a comparison of the phylogenetic and biosynthetic affiliations of terrestrial and marine natural products and their sources.The “taxonomic chemistry tree” in Fig. 5 illustrates the resemblance between marine invertebrate and microbial compounds. The tree, organized phylogenetically by source, shows a variety of natural products that are presumably produced by PKS and NRPS pathways. The tree illustrates natural products from a variety of marine organisms, highlighting compounds isolated from marine macroorganisms versus terrestrial analogues produced entirely by microorganisms. In some cases the compounds are identical, such as westiellamide6717 and cycloxazoline6818 found in a terrestrial cyanobacterium and marine ascidian, respectively. More commonly, there are specific functional groups or skeletal systems that are similar, for example: the enediyne “warhead” moiety in calicheamicin6919 and namenamicin7020, the isoquinoline ring system of saframycin B7121 and ecteinascidin-74372,7322, and the overall structural analogies between nodularin7423 and motuporin7524.
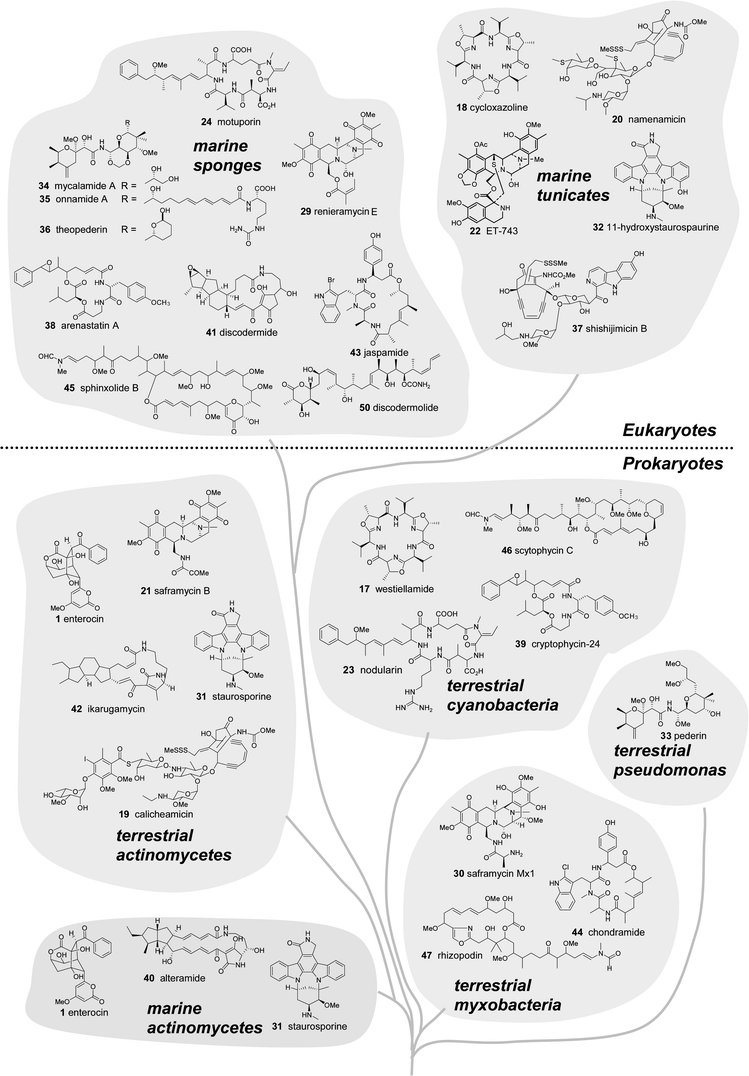 | ||
| Fig. 5 Marine natural products and structural analogues from terrestrial microbes arranged in a “chemo-taxonomic tree”. Compounds isolated from prokaryotes (bacteria) are shown in the lower half and metabolites from sponges and ascidians (multicellular eukaryotes) are shown in the upper half of the diagram. | ||
5.1 Limitations of cellular localization studies
There have been a number of studies directed at elucidating the biosynthetic sources of marine natural products.62,76–78 The most unequivocal evidence to demonstrate a symbiont origin of secondary metabolites is to culture the microbe and show production of the compound. However, despite recent advances in the culturing of new marine bacteria, there are few examples of an invertebrate-associated microbe in culture that produces the original metabolite.36,79 The lack of progress in this area could be due to sub-optimal isolation and selection methodologies, the inability of the bacteria to produce the compounds under the given growth conditions or the need for a mixed culture of microbes (and perhaps host cells) to biosynthesize the metabolite.80 Since these strategies have failed to identify symbionts that produce the original “invertebrate” compounds, other culture-independent methods have been employed to examine this hypothesis.58,78,81 One common strategy that has been used in recent years is the localization of compounds in specific invertebrate or symbiont cells (reviewed elsewhere in this issue by Haygood et al.).Unson and coworkers isolated the poly-brominated biphenyl ether 25 in the cyanobacterial symbiont Oscillatoria spongeliae separated from the sponge Dysidea herbacea.82 The same group also localized the chlorinated amino acid derivative 13-demethylisodysidolide 12 from O. spongeliae separated from another specimen of D. herbacea.58 The cyclic peptide theopalauamide 26 was isolated from filamentous δ-proteobacteria separated from the sponge Theonella swinhoei while the cytotoxic polyketide swinholide A 27 was isolated from the fraction containing a mixture of unicellular bacteria from the same sponge.63,81 The polyketide latrunculin B 28 was localized in situ within sponge cells of Negombata magnifica using gold-labeled anti-latrunculin antibodies.76
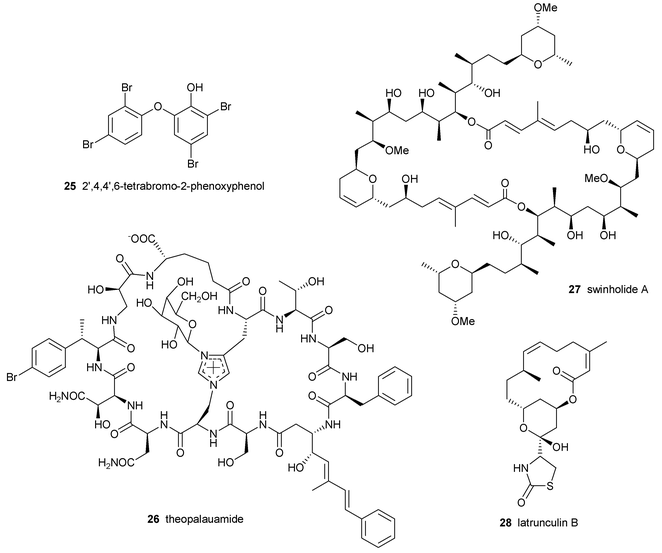
While the above examples suggest that the compounds are associated with specific bacteria or invertebrate cells, the possibility that the compounds were synthesized in another cell type and translocated to those storage cells cannot be ruled out. Theoretically, if a sponge or ascidian has evolved to harbor a microbial symbiont that produces a useful metabolite, it could be advantageous for the invertebrate to adsorb the compound onto its cell surfaces to prevent fouling or uptake it into specific cells for storage and protection from predation, for example.83 Virtually nothing is known about the transport of natural products or their precursors between invertebrate cells and bacteria. Terrestrial bacteria are especially well known for their ability to excrete natural products into the environment as exemplified by self resistance export mechanisms and the extra-cellular occurrence of many antibiotics produced in fermentation such as penicillin, streptomycin and mitomycin C.84,85
It is absolutely relevant to study where and how secondary metabolites are stored to understand their roles in the ecology and biology of the invertebrates and/or their associated microbes. However, the only ways to determine the true source of secondary metabolites are to culture the symbiont and demonstrate compound production or utilize the molecular genetic tools that are now available to identify and locate the biosynthetic clusters.
6 Molecular genetic approaches
The investigation of secondary metabolism using molecular genetics has the potential to provide biosynthetic information that can be used in different ways. First, the cluster sequences can be compared to others in extensive online databases to identify related genes and their source organisms. Second, compound-specific genes combined with 16S rRNA probes can be used to identify and localize the source of the cluster using in situ hybridization. Third, the identification of likely microbial sources can direct efforts to better replicate appropriate conditions to obtain the producing microorganism in culture.The major steps involved in developing an integrated biosynthetic study to obtain the gene cluster and source information are shown in Fig. 6. The first step in a search for any specific gene cluster is the prediction of the biosynthetic pathway based on our growing knowledge of other modular systems. This analysis is critical to identify specific, key enzymes that may be used as molecular probes to screen genomic libraries.
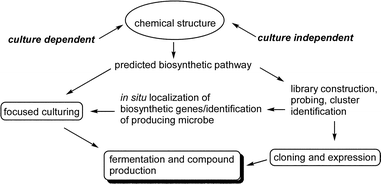 | ||
| Fig. 6 Overview of a molecular genetic/cloning strategy for marine compounds. | ||
6.1 Marine compounds with terrestrial analogues (known gene cluster)
There are currently several examples of identified gene clusters from terrestrial microbes that encode compounds similar or identical to those from marine sources (Table 2). A detailed analysis of the terrestrial clusters is beyond the scope of this review, but in Table 2 we present a brief description of each suggested pair of compounds, their source organisms and biosynthetic pathways. The availability of analogous genes/clusters makes these marine compounds the most tractable targets for molecular genetic studies. Given the clustering and role of horizontal transfer of genes in prokaryotes, it is unlikely that the genes encoding the same secondary metabolite in the two organisms could arise separately by the process of convergent evolution, but cannot be ruled out.105,106 Another appealing suggestion would involve the origination from a common microbial ancestor, with new chemical variations arising from the processes of genetic selection, genetic drift or perhaps speciation resulting from environmental separation.104,107| Terrestrial compound | Microbial source(s) | Biosynthetic pathway | Related marine compound | Microbial/invertebrate sources |
|---|---|---|---|---|
| staurosporine8631 | Streptomyces staurosporeus | Shikimic acid87 | staurosporine8831 11-hydroxystaurosporine8932 | marine actinomycete ascidian (Eudistoma sp.) |
| saframycin Mx190,9130 saframycin B9221 | Myxococcus xanthus Streptomyceslavendulae | NRPS93 | renieramycin E9429 ET-74372,7322 | sponge Reniera sp. ascidian Ecteinascidia turbinata |
| pederin9533 | Endosymbiont of the Paederus blister beetle, most similar to Pseudomonas aeruginosa | Mixed type I PKS/NRPS96 | mycalamide A9734 onnamide A9835 theopederin9936 | sponge (Mycale sp.) sponge (Theonella sp.) sponge (Theonella sp.) |
| calicheamicin6919 | Micromonospora echinospora | Iterative type I PKS100,101 | namenamicin7020 shishijimicin B10237 | ascidian (Polysyncraton lithostrotum) ascidian (Didemnum proliferum) |
| nodularin7423 | Microcystis aeruginosa Planktothrix agardhii | Mixed type I PKS/NRPS103,104 | motuporin7524 | sponge Theonella swinhoei |
One example that deserves further discussion is the potent anti-tumor alkaloid ET-743 22. The ecteinascidins (ETs) are a group of bioactive tetrahydroisoquinoline metabolites that were isolated from the Caribbean ascidian Ecteinascidia turbinata in low yields.72,73 ET-743 is the most bioactive analogue and is one of the most clinically advanced marine natural products. The complex core structure bears remarkable similarity to several sponge compounds (i.e. renieramycin 29) and terrestrial microbial metabolites such as saframycin B 21 and saframycin Mx1 30 isolated from Streptomyceslavendulae92 and Myxococcus xanthus, respectively.90,91 Currently the compound is supplied by a relatively efficient semi-synthetic strategy starting with the microbially produced cyanosafracin B obtained from Pseudomonas fluorescens.108
Despite the synthetic accessibility of ET-743, there is interest in identifying and isolating a presumed microbial symbiont from the ascidian that produces the compound in culture. A study by Moss et al. utilized 16S rDNA libraries, in situ hybridization and microscopy to demonstrate the consistent association of the ascidian larvae with a γ-proteobacterium denoted ‘Candidatus Endoecteinascidia frumentensis’.109 Additional circumstantial support for microbial production of ET-743 in the ascidian is preliminary work showing the consistent presence of both the ecteinascidin compounds and a single major strain of bacteria in the larval tissue.110
Biosynthetic feeding studies of the saframycins/ecteinascidin compounds have shown that the core structure is formed by the condensation of two tyrosine derivatives via a diketopiperazine intermediate and the incorporation of glycine and alanine.111,112 Additionally, the methyl groups are known to be derived from S-adenosyl methionine. Cysteine was also found to be incorporated into the ecteinascidins.112 The saframycin Mx1 gene cluster is in good agreement with the precursor studies, encoding two NRPS proteins that incorporate the successive condensation of AlaGlyTyrTyr as specified by the 4 adenylation domains. An O-methyl transferase was also found to be necessary for saframycin Mx1 production. Although the ecteinascidins possess additional functional groups (exocyclic isoquinoline and sulfur bridge), the genetic structure of the core ET pathway provides the sequence information necessary to probe the ascidian metagenome to potentially identify the biosynthetic source of the ecteinascidin compounds.93
6.2 Marine compounds with terrestrial analogues (no known gene cluster)
For marine compounds with close terrestrial analogues that have no known gene cluster available, it may be worthwhile to focus first on obtaining specific molecular probes and preliminary sequence information from the pure terrestrial microbial culture. Some examples of marine natural products with terrestrial analogues are shown in Table 3.| Marine compound | Microbial/invertebrate sources | Proposed biosynthetic pathway | Terrestrial compound | Microbial source(s) |
|---|---|---|---|---|
| arenastatin11338 | sponge (Dysidea arenaria) | Mixed type I PKS/NRPS | cryptophycin-2411439 | Nostoc sp. |
| alteramide11540 discodermide11641 | bacterium (Alteromonas sp.) sponge (Discodermia dissoluta) | Mixed type I PKS/NRPS | ikarugamycin11742 | Streptomyces sp. |
| jaspamide11843 | sponge (Jaspis splendins) | Mixed type I PKS/NRPS | chondramide11944 | Chondromyces crocatus |
| sphinxolide12045 | sponge (Neosiphonia superstes) | PKS | scytophycin12146 | Scytonema pseudohofmanni |
| sphinxolide12045 | sponge (Neosiphonia superstes) | PKS | rhizopodin12247 | Myxococcus stipitatus |
One model system in this category includes the Dysidea sponge compound arenastatin11338, identical to cryptophycin-2411439 produced as a minor compound by terrestrial Nostoc cyanobacteria. Interestingly, one of the strains that produces the cryptophycins, Nostoc sp. 53789, was isolated as the symbiont of a lichen.123 The Nostoc cyanobiont in this case is much more tractable because it is available in pure culture, can be easily grown in the laboratory for unlimited sources of DNA, and may be amenable to genetic manipulations. The sponge, however, must be obtained by scuba, cannot be grown in a lab, and contains diverse populations of bacteria.124 Identification of the cryptophycin gene cluster first could provide specific probes to localize the presumably similar cluster in the sponge. Additionally, the similarity in structures suggests that the sponge symbiont responsible for arenastatin production is probably also a cyanobacterium and this information is currently directing efforts in our lab to culture the producing organism.125
6.3. Marine compounds with no known terrestrial analogues
A majority of marine natural products appear to be unique, with no known obvious structural analogues from terrestrial sources, such as okadaic acid 48 and bryostatin 49. However, analysis of such structures can lead to a probable PKS and/or NRPS pathway, and therefore, likely microbial origin. These compounds are potentially tractable by molecular genetics studies due to their presumed production by modular enzymes, and may be accessed using the same strategy shown in Fig. 6.
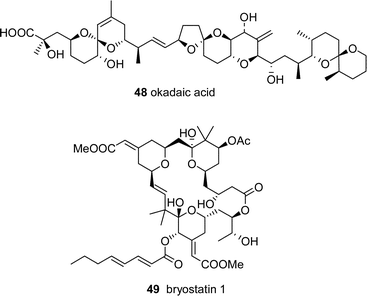
7 Prediction of marine natural product pathways
To demonstrate how a metagenomic approach could be designed for compounds with no known terrestrial analogues from even more complex assemblages, examples of predicted biosynthetic “maps” are given for the polyketide discodermolide 50 and the nonribosomally synthesized didemnin B 51 isolated from a sponge and ascidian, respectively. These compounds were chosen for analysis due to their potent bioactivities, lack of economically efficient syntheses, probability of a symbiont producer and need for alternatives to wild collection of the invertebrates.
The proposed PKS gene cluster for the biosynthesis of discodermolide 50 and analogues 52–56 is shown in Fig. 7. The predicted starter unit is acetyl-CoA which is presumably elongated by 4 malonate and 7 methyl malonate subunits. A unique feature of the biosynthesis is that it includes three trans–cis isomerizations that may occur during the reductive loop of the β-carbonyl through the stereochemically specific reduction by the KR and subsequent dehydration or possibly by a separate isomerase. Another unprecedented component of the proposed biosynthetic pathway is the predicted formation of the 8,9 cis double bond by dehydration in the direction of the growing chain, rather than towards the “starting” end (indicated by * in Fig. 7). Presumably the final linear chain undergoes an intramolecular cyclization to form the terminal 6-membered lactone ring and is simultaneously cleaved from the protein complex by a terminal thioesterase (TE) domain. Tailoring enzymes, including a C-methyltransferase and carbamoyl transferase may then elaborate the core structure to add a methyl at C-3 and carbamate moiety to the hydroxyl at C-19, respectively. A minor component, 9(13)-cyclodiscodermolide 56, may be formed by an intramolecular C–C bond formation by a cation–π cyclization (typical in terpene cyclases) to form the new 5-membered ring.
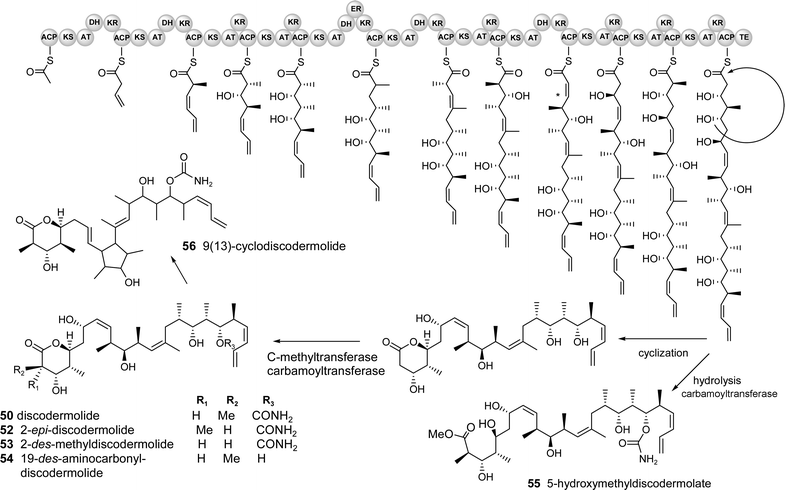 | ||
| Fig. 7 Predicted biosynthetic pathway for discodermolide and derivatives. (The cluster probably contains several genes but the organisation/number of modules cannot be predicted and it is therefore depicted as a single gene for simplicity) | ||
One of the technical challenges associated with a search for this putative cluster is that the sponge is presumably associated with many different microorganisms, both dietary and symbiotic, and may require an intractable number of cosmid clones to adequately cover all of the possible genomes present (including that of the eukaryotic sponge, estimated to be 1.7 Gb in some species based on cellular DNA content).132 One approach to reduce the size of a potentially unmanageable genomic library for suspected sponge symbionts is to separate the major populations of microbes from the sponge, shown to be feasible in the previously discussed cellular localization studies. Bacterial enrichment can be accomplished with dissociated cells by fluorescence activated cell sorting (FACS, for photosynthetic organisms),58,82 centrifugation and/or filtration,81 and density gradients.133 An alternative metagenomic approach was recently used successfully by Piel to isolate the PKS gene cluster encoding the toxin pederin 33 from a mixture of terrestrial beetle tissue and one major species of symbiotic bacteria.96 A large cosmid library was constructed (80,000 clones) and a rapid PCR based screen using a “pooling” method was used to quickly isolate the positive clones.96 Although the beetle was previously shown to possess only a single symbiont, far fewer than any marine sponge, such a metagenomic strategy may be successful if probes are carefully designed to be specific for the pathway of interest. In this example for discodermolide, a combination of ketosynthase domains, C-methyl transferase genes and the carbamoyl transferase could be used to target the cluster. The carbamoyl transferase would be a particularly good handle because it produces a rare functional group, present only in a few microbial natural products such as novobiocin,134a bleomycin,134b cephamycin C134c and mitomycin C.134d
The proposed NRPS gene cluster for the biosynthesis of didemnin B/dehydrodidemnin B is shown in Fig. 8. This retro-biosynthetic analysis takes into consideration known progenitors, which point to the suggestion that N-methyl leucine serves as the starter unit for the NRPS assembly-line. Didemnin A, found in the highest yield from the Caribbean tunicate, contains only the core peptolide (no side-chain), suggesting that this side-chain is not integral to the main didemnin B assembly line. From this start point, a series of extensions of proteinogenic (L-leucine, L-proline and threonine), nonproteinogenic amino acids (isostatine, N-methyl tyrosine, N-methyl leucine) and a fatty acid component (hydroxylvaleryl isopropionate) are predicted to occur. Following the build-up of the assembly line a thioesterase enzyme is predicted to catalyze the release of the enzyme thioester-linked heptadepsipeptide coupled with a macrocyclization reaction. This reaction would generate the didemnin A structure that may serve as a substrate for post core-assembly enzymes with varying specificities for side-chain groups. From this proposed molecular assembly sequence, the known diversity of the didemnin family of marine natural products could conceivably arise from one core biosynthetic system (Fig. 8).
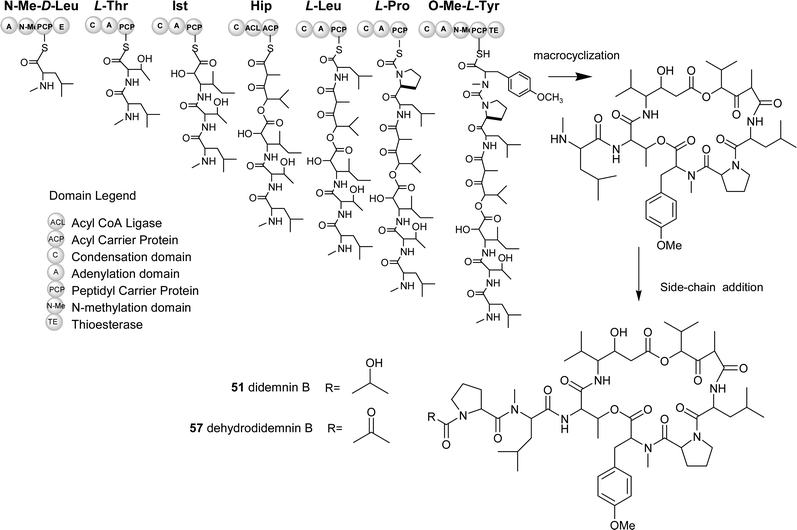 | ||
| Fig. 8 Predicted biosynthetic pathway for didemnin B and derivatives. (NMe-D-Leu: N-methyl-D-Leucine, Ist: Isostatine, Hip: Hydroxylvaleryl isopropionyl, O-Me-L-Tyr: O-methyl-L-Tyrosine) | ||
The recent advances in nonribosomal peptide biosynthesis and methodologies to generate modifications of these pathways make biologically active marine depsipeptides like the didemnins particularly attractive targets. A guided search for the didemnin B genes could involve utilizing the inherent co-linearity rules for nonribosomal peptide biosynthesis coupled with analysis of the nonribosomal code.137,138 An example of a feasible approach would follow a random genetic amplification and in silico study of A (adenylation) domains obtained using metagenomic template DNA from the tunicate. Such a strategy would afford a rapid snap-shot of all possible NRPS “gatekeeper” domains (A domains) from the tunicate. More importantly, this approach would allow for honing in on particular chemical functional groups present in the natural didemnins. One particular chemical handle is the N-methyl leucine and its relative position in these marine natural products. From the pool of A domains, an in silico candidate would be selected on the basis of having a nonribosomal code specific for leucine (Aleu). Following this, libraries containing material enriched for bacterial DNA would be screened using this specific domain. Once positive clones were identified using the predicted Aleu sequences, a genetic walk (DNA sequencing) immediately upstream and downstream could provide additional cluster information. If the “downstream” sequences revealed an embedded N-methyltransferase domain, it is plausible that the cloned DNA relates to didemnin B biosynthesis. Information gathered from peptide antibiotic clusters has established that well known N-methylations of amide bonds in peptide antibiotics occur by such embedded domains.137 To complete the cluster, the entire cosmid DNA would be sequenced and the resulting sequences checked against a pathway prediction such as the one shown in Fig. 8.
8 Identification and culturing of microbial symbionts
There are two major routes for identifying presumed symbiotic sources from invertebrates using biosynthetic information. A first approach utilizes a gene cluster localization strategy. Once the putative genes/cluster is identified for a metabolite, this information can be used to localize the genes to a specific bacterium using in situ mRNA hybridization (demonstrated in the bryostatin example, above). The identity of the microbes can simultaneously be probed by identifying all of the associated bacteria present by 16S rDNA libraries and/or DGGE (denaturing gradient gel electrophoresis) and then using 16S rRNA probes to localize group- or species-specific signals to the microbial cells.139,140 A second method is to simply utilize the phylogenetic and culturing information known from organisms that produce similar chemistry. For example, with the sponges that produce the pederin-like compounds (34–36), culturing efforts could focus on conditions specifically related to pseudomonads. Similarly, the motuporin 24 producing sponge could be approached using conditions specific for marine cyanobacteria. Even though known microbial symbionts can be notoriously difficult to culture, we believe that recent, directed microbiology efforts point to a potentially much greater number of culturable marine bacteria from invertebrates and the marine environment in general.37,125,1409 Will marine microbial diversity translate to chemical richness?
Stemming from early investigations of terrestrial plants, the utility of natural products as therapeutic agents was realized. From these studies, the hypothesis was developed that organismal diversity correlates with the structural variety of chemical compounds. Continuous explorations of plant secondary metabolites led to strong support for this theory which developed into a paradigm for natural products research. The discovery of the propensity of soil dwelling actinomycetes (especially Streptomyces) to produce antibiotics led to a transformation of natural product-based drug discovery.141 The chemical investigations of the actinomycetales stimulated the exploration of a wide range of microorganisms that were shown to also produce antibiotics and other biologically active compounds.142 The diversity of terrestrial microbial compounds is unparalleled and we now know that this is directly related to the modularity of secondary metabolic systems (i.e. polyketide and nonribosomal peptide pathways).The paradigm of biodiversity translating to chemodiversity is also clearly evident from explorations of natural products from organisms in the marine environment.35 Soft bodied invertebrates, in particular, have yielded a wide variety of biologically active structures. From an ecological standpoint, such organisms may have evolved the ability to harbor or produce chemicals (such as defensive or allelopathic compounds) that promote their overall fitness. The similarity of many “invertebrate” compounds to those isolated from terrestrial microbes (Fig. 5) led to the hypothesis that associated microorganisms might be responsible for their production. Sessile invertebrates such as sponges and ascidians are especially known to associate with diverse populations of bacteria. In cases where there are clear parallels between marine and terrestrial compounds (i.e. arenastatin and cryptophycin) it is logical to infer that the marine metabolite is produced by an associated symbiont. In the majority of other peptide and polyketide compounds with no known terrestrial analogues, it is possible to invoke a feasible modular assembly system. These complex biosynthetic enzymes (PKS and NRPS) are only known to be encoded by the genomes of microorganisms.
Studies have shown that marine invertebrates house extremely high levels of microbial diversity and are a logical source for culturing and molecular genetic studies.36,115,143–146 Current research suggests that unique microorganisms are also abundant in all areas of the ocean, and two new actinomycete genera have been recently suggested.37,125 More importantly, these new organisms are being investigated for their production of new compounds, and at least one published study of compounds isolated from the newly proposed genus Salinospora has led to the elucidation of novel bioactive metabolites.147 The number of unique natural products from cultured marine microbes with promising biological activity is growing and may translate to a new resource for drug discovery.40
10 Future prospects
The inherent difficulties associated with culturing symbionts implicated in the production of specific compounds have stymied efforts to develop sustainable sources of valuable marine “invertebrate” metabolites. However, the growing number of biosynthetic clusters identified and cloned from culturable marine bacteria may soon pave the way for similar studies in symbionts in more complex associations. Additionally, recent pioneering efforts in marine microbiology are demonstrating the isolation of unique, diverse microbes from all areas of the ocean environment using directed culturing methods. This growing source of novel metabolites is rapidly becoming the focus of new molecular genetic studies and may provide even more diverse enzymatic activities for the combinatorial “toolbox”.We expect that the combined knowledge of microbial biodiversity and the ability to target specific biosynthetic genes will provide the necessary tools to identify and culture microbes that produce novel, potent pharmaceuticals on an economically viable scale. The decreasing costs associated with whole genome sequencing will also allow for a much greater number of bacterial and possibly invertebrate genomes to be sequenced, providing a genetic basis to identify and clone new pathways. Thus, the synergistic efforts of both biological and chemical approaches will provide insight into the biosynthesis and eventual production of some of the most structurally complex, bioactive marine natural products.
One thing that we have learned from terrestrial systems is that the search for specific genes in an organism or complex assemblage not only sets the stage for exploring natural diversity but provides a framework for understanding biosynthesis at its most fundamental level. Natural products discovery has greatly benefited from the investigations of the marine environment and the understanding of the roles that secondary metabolites may play in the ecology of marine organisms. Moving forward and tapping into the diversity and metabolic talents of the vastly under-explored world of marine microorganisms will undoubtedly provide a barrage of new natural products and should lead to high quality bioactive drug leads. In a greater sense, this type of investigative work could revitalize drug discovery from nature and provide us with the genetic storehouse of PKS and NRPS enzymes for understanding the roles and true potential of secondary metabolites from marine ecosystems.
11 Acknowledgements
Investigations of marine microbial derived gene clusters in the laboratory of DHS are funded by P01 CA 83155. CES is supported by a MinnCResT NIH postdoctoral training fellowship. We thank John Faulkner for being a wonderful inspiration and mentor to many young scientists interested in studying the biosynthesis of marine natural products.12 References
- ISI Web of Knowledge online reference database 2003 http://isi3.newisiknowledge.com/portal.cgi.
- PubMed online database 2003 http://www.ncbi.nlm.nih.gov/.
- D. E. Cane, F. Kudo, K. Kinoshita and C. Khosla, Chem. Biol., 2002, 9, 131–142 CrossRef CAS.
- B. J. Beck, C. C. Aldrich, R. A. Fecik, K. A. Reynolds and D. H. Sherman, J. Am. Chem. Soc., 2003, 125, 4682–4683 CrossRef CAS.
- J. Piel, K. Hoang and B. S. Moore, J. Am. Chem. Soc., 2000, 122, 5415–5416 CrossRef CAS.
- A. Y. Li and J. Piel, Chem. Biol., 2002, 9, 1017–1026 CrossRef CAS.
- Z. X. Chang, P. Flatt, W. H. Gerwick, V. A. Nguyen, C. L. Willis and D. H. Sherman, Gene, 2002, 296, 235–247 CrossRef CAS.
- R. V. Snyder, P. D. L. Gibbs, A. Palacios, L. Abiy, R. Dickey, J. V. Lopez and K. S. Rein, Mar. Biotechnol., 2003, 5, 1–12 CrossRef CAS.
- M. C. Moffitt and B. A. Neilan, J. Mol. Evol., 2003, 56, 446–457 CrossRef CAS.
- S. K. Davidson, S. W. Allen, G. E. Lim, C. M. Anderson and M. G. Haygood, Appl. Environ. Microbiol., 2001, 67, 4531–4537 CrossRef CAS.
- University of Canterbury. MarinLit Database. 2002.
- D. J. Faulkner, Ant. Van Leeuw. Int. J. Gen. Mol. Microbiol., 2000, 77, 135–145 Search PubMed.
- B. Haefner, Drug Discovery Today, 2003, 8, 536–544 CrossRef CAS.
- D. J. Newman and G. M. Cragg, Curr. Med. Chem., 2003, in press Search PubMed.
- A. M. S. Mayer and K. R. Gustafson, Int. J. Cancer, 2003, 105, 291–299 CrossRef CAS.
- K. L. Rinehart, J. B. Gloer, R. G. Hughes, H. E. Renis, J. P. Mcgovren, E. B. Swynenberg, D. A. Stringfellow, S. L. Kuentzel and L. H. Li, Science, 1981, 212, 933–935 CAS.
- M. H. G. Munro, J. W. Blunt, E. J. Dumdei, S. J. H. Hickford, R. E. Lill, S. X. Li, C. N. Battershill and A. R. Duckworth, J. Biotechnol., 1999, 70, 15–25 CrossRef CAS.
- A. L. Demain, Biotechnol. Adv., 2000, 18, 499–514 CrossRef CAS.
- L. K. Xiang, J. A. Kalaitzis, G. Nilsen, L. Chen and B. S. Moore, Org. Lett., 2002, 4, 957–960 CrossRef CAS.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- C. Khosla, Chem. Rev., 1997, 97, 2577–2590 CrossRef CAS.
- D. A. Hopwood, Chem. Rev., 1997, 97, 2465–2497 CrossRef CAS.
- D. Schwarzer, R. Finking and M. A. Marahiel, Nat. Prod. Rep., 2003, 20, 275–287 RSC.
- T. Weber and M. A. Marahiel, Structure, 2001, 9, R3–R9 CrossRef CAS.
- D. Schwarzer and M. A. Marahiel, Naturwissenschaften, 2001, 88, 93–101 CrossRef CAS.
- D. A. Hopwood and D. H. Sherman, Annu. Rev. Gen., 1990, 24, 37–66 Search PubMed.
- M. B. Austin and A. J. P. Noel, Nat. Prod. Rep., 2003, 20, 79–110 RSC.
- B. S. Moore and C. Hertweck, Nat. Prod. Rep., 2002, 19, 70–99 RSC.
- U. Rix, C. Fischer, L. L. Remsing and J. Rohr, Nat. Prod. Rep., 2002, 19, 542–580 RSC.
- H. D. Mootz, D. Schwarzer and M. A. Marahiel, Chembiochem, 2002, 3, 491–504 CrossRef CAS.
- D. P. Mankelow and B. A. Neilan, Expert Opin. Ther. Patents, 2000, 10, 1583–1591 Search PubMed.
- T. Stachelhaus and M. A. Marahiel, FEMS Microbiol. Lett., 1995, 125, 3–14 CrossRef CAS.
- C. T. Walsh, H. W. Chen, T. A. Keating, B. K. Hubbard, H. C. Losey, L. S. Luo, C. G. Marshall, D. A. Miller and H. M. Patel, Curr. Opin. Chem. Biol., 2001, 5, 525–534 CrossRef CAS.
- R. M. Morris, M. S. Rappe, S. A. Connon, K. L. Vergin, W. A. Siebold, C. A. Carlson and S. J. Giovannoni, Nature, 2002, 420, 806–810 CrossRef CAS.
- A. T. Bull, A. C. Ward and M. Goodfellow, Microb. Mol. Biol. Rev., 2000, 64, 573–606 Search PubMed.
- P. R. Jensen and W. Fenical, J. Ind. Microb. Biotechnol., 1996, 17, 346–351 Search PubMed.
- T. J. Mincer, P. R. Jensen, C. A. Kauffman and W. Fenical, Appl. Environ. Microbiol., 2002, 68, 5005–5011 CrossRef CAS.
- J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2003, 20, 1–48 RSC.
- J. Piel, C. Hertweck, P. R. Shipley, D. M. Hunt, M. S. Newman and B. S. Moore, Chem. Biol., 2000, 7, 943–955 CrossRef CAS.
- V. S. Bernan, M. Greenstein and W. M. Maiese, in Adv. Appl. Microbiol., Academic Press, Orlando, 1997, pp. 57–90 Search PubMed.
- N. Miyairi, H. I. Sakai, T. Konomi and H. Imanaka, J. Antibiot., 1976, 29, 227–235 CAS.
- N. Sitachitta, M. Gadepalli and B. S. Davidson, Tetrahedron, 1996, 52, 8073–8080 CrossRef CAS.
- H. Kang, P. R. Jensen and W. Fenical, J. Org. Chem., 1996, 61, 1543–1546 CrossRef CAS.
- N. Sitachitta, M. Gadepalli and B. S. Davidson, Tetrahedron, 1996, 52, 8073–8080 CrossRef CAS.
- P. Babczinski, M. Dorgerloh, A. Lobberding, H. J. Santel, R. R. Schmidt, P. Schmitt and C. Wunsche, Pestic. Sci., 1991, 33, 439–446 Search PubMed.
- L. K. Xiang and B. S. Moore, J. Biol. Chem., 2002, 277, 32505–32509 CrossRef CAS.
- L. K. Xiang and B. S. Moore, J. Bacteriol., 2003, 185, 399–404 CrossRef CAS.
- H. Seto, T. Sato, S. Urano, J. Uzawa and H. Yonehara, Tetrahedron Lett., 1976, 4367–4370 CrossRef.
- J. L. C. Wright, T. Hu, J. L. McLachlan, J. Needham and J. A. Walter, J. Am. Chem. Soc., 1996, 118, 8757–8758 CrossRef CAS.
- M. E. Goldman, G. S. Salituro, J. A. Bowen, J. M. Williamson, D. L. Zink, W. A. Schleif and E. A. Emini, Mol. Pharmacol., 1990, 38, 20–25 Search PubMed.
- M. Chino, K. Nishikawa, T. Tsuchida, R. Sawa, H. Nakamura, K. T. Nakamura, Y. Muraoka, D. Ikeda, H. Naganawa, T. Sawa and T. Takeuchi, J. Antibiot., 1997, 50, 143–146 CAS.
- T. Ueno, H. Takahashi, M. Oda, M. Mizunuma, A. Yokoyama, Y. Goto, Y. Mizushina, K. Sakaguchi and H. Hayashi, Biochemistry, 2000, 39, 5995–6002 CrossRef CAS.
- E. Dittmann, B. A. Neilan and T. Borner, Appl. Microb. Biotechnol., 2001, 57, 467–473 Search PubMed.
- M. C. Moffitt and B. A. Neilan, FEMS Microbiol. Lett., 2001, 196, 207–214 CrossRef CAS.
- N. A. Magarvey, D. Abelson and D. H. Sherman, 2003, manuscript in preparation.
- M. D. Unson, C. B. Rose, D. J. Faulkner, L. S. Brinen, J. R. Steiner and J. Clardy, J. Org. Chem., 1993, 58, 6336–6343 CrossRef CAS.
- J. B. MacMillan and T. F. Molinski, J. Nat. Prod., 2000, 63, 155–157 CrossRef CAS.
- M. D. Unson and D. J. Faulkner, Experientia, 1993, 49, 349–353 Search PubMed.
- J. Orjala and W. H. Gerwick, J. Nat. Prod., 1996, 59, 427–430 CrossRef CAS.
- J. I. Jimenez and P. J. Scheuer, J. Nat. Prod., 2001, 64, 200–203 CrossRef CAS.
- N. Sitachitta, B. L. Marquez, R. T. Williamson, J. Rossi, M. A. Roberts, W. H. Gerwick, V. A. Nguyen and C. L. Willis, Tetrahedron, 2000, 56, 9103–9113 CrossRef CAS.
- D. J. Faulkner, H. Y. He, M. D. Unson, C. A. Bewley and M. J. Garson, Gazz. Chim. Ital., 1993, 123, 301–307 CAS.
- E. W. Schmidt, A. Y. Obraztsova, S. K. Davidson, D. J. Faulkner and M. G. Haygood, Mar. Biol., 2000, 136, 969–977 CrossRef CAS.
- C. A. Bewley and D. J. Faulkner, Angew. Chem., 1998, 37, 2163–2178 CAS.
- B. S. Moore, Nat. Prod. Rep., 1999, 16, 653–674 RSC.
- R. Kerr, A. Kohl, J. Lopez, PCT Int. Appl. WO 2003065001, 8-7-2003.
- M. R. Prinsep, R. E. Moore, I. A. Levine and G. M. L. Patterson, J. Nat. Prod., 1992, 55, 140–142 CrossRef CAS.
- T. W. Hambley, C. J. Hawkins, M. F. Lavin, A. Vandenbrenk and D. J. Watters, Tetrahedron, 1992, 48, 341–348 CrossRef CAS.
- M. D. Lee, T. S. Dunne, C. C. Chang, G. A. Ellestad, M. M. Siegel, G. O. Morton, W. J. Mcgahren and D. B. Borders, J. Am. Chem. Soc., 1987, 109, 3466–3468 CrossRef CAS.
- L. A. McDonald, T. L. Capson, G. Krishnamurthy, W. D. Ding, G. A. Ellestad, V. S. Bernan, W. M. Maiese, P. Lassota, C. Discafani, R. A. Kramer and C. M. Ireland, J. Am. Chem. Soc., 1996, 118, 10898–10899 CrossRef CAS.
- T. Arai, K. Takahashi, A. Kubo, S. Nakahara, S. Sato, K. Aiba and C. Tamura, Tetrahedron Lett., 1979, 2355–2358 CrossRef CAS.
- K. L. Rinehart, T. G. Holt, N. L. Fregeau, J. G. Stroh, P. A. Keifer, F. Sun, L. H. Li and D. G. Martin, J. Org. Chem., 1990, 55, 4512–4515 CrossRef CAS.
- A. E. Wright, D. A. Forleo, G. P. Gunawardana, S. P. Gunasekera, F. E. Koehn and O. J. McConnell, J. Org. Chem., 1990, 55, 4508–4512 CrossRef CAS.
- K. L. Rinehart, K. Harada, M. Namikoshi, C. Chen, C. A. Harvis, M. H. G. Munro, J. W. Blunt, P. E. Mulligan, V. R. Beasley, A. M. Dahlem and W. W. Carmichael, J. Am. Chem. Soc., 1988, 110, 8557–8558 CrossRef.
- E. D. Desilva, D. E. Williams, R. J. Andersen, H. Klix, C. F. B. Holmes and T. M. Allen, Tetrahedron Lett., 1992, 33, 1561–1564 CrossRef.
- O. Gillor, S. Carmeli, Y. Rahamim, Z. Fishelson and M. Ilan, Mar. Biotechnol., 2000, 2, 213–223 CAS.
- C. E. Salomon and D. J. Faulkner, J. Nat. Prod., 2002, 65, 689–692 CrossRef CAS.
- J. E. Thompson, K. D. Barrow and D. J. Faulkner, Acta Zool., 1983, 64, 199–210.
- B. S. Davidson, Curr. Opin. Biotechnol., 1995, 6, 284–291 CrossRef CAS.
- M. Cueto, P. R. Jensen, C. Kauffman, W. Fenical, E. Lobkovsky and J. Clardy, J. Nat. Prod., 2001, 64, 1444–1446 CrossRef CAS.
- C. A. Bewley, N. D. Holland and D. J. Faulkner, Experientia, 1996, 52, 716–722 Search PubMed.
- M. D. Unson, N. D. Holland and D. J. Faulkner, Mar. Biol., 1994, 119, 1–11 CAS.
- M. E. Hay, J. Exp. Mar. Biol. Ecol., 1996, 200, 103–134 CrossRef CAS.
- R. Pratt and J. Dufrenoy, Antibiotics, 2nd edn.; Lippincott Co., Philadelphia, 1953 Search PubMed.
- P. J. Sheldon, D. A. Johnson, P. R. August, H. W. Liu and D. H. Sherman, J. Bacteriol., 1997, 179, 1796–1804 CAS.
- A. Furusaki, N. Hashiba, T. Matsumoto, A. Hirano, Y. Iwai and S. Omura, J. Chem. Soc., Chem. Commun., 1978, 800–801 RSC.
- H. Onaka, S. Taniguchi, Y. Igarashi and T. Furumai, J. Antibiot., 2002, 55, 1063–1071 CAS.
- D. E. Williams, V. S. Bernan, F. V. Ritacco, W. M. Maiese, M. Greenstein and R. J. Andersen, Tetrahedron Lett., 1999, 40, 7171–7174 CrossRef CAS.
- R. B. Kinnel and P. J. Scheuer, J. Org. Chem., 1992, 57, 6327–6329 CrossRef CAS.
- H. Irschik, W. Trowitzschkienast, K. Gerth, G. Hofle and H. Reichenbach, J. Antibiot., 1988, 41, 993–998 CAS.
- W. Trowitzschkienast, H. Irschik, H. Reichenbach, V. Wray and G. Hofle, Liebigs Ann. Chem., 1988, 475–481 Search PubMed.
- T. Arai, K. Takahashi and A. Kubo, J. Antibiot., 1977, 30, 1015–1018 CAS.
- A. Pospiech, B. Cluzel, J. Bietenhader and T. Schupp, Microbiol. UK, 1995, 141, 1793–1803 Search PubMed.
- J. M. Frincke and D. J. Faulkner, J. Am. Chem. Soc., 1982, 104, 265–269 CrossRef CAS.
- A. Fursaki, T. Watanabe, T. Matsumoto and M. Yanagiya, Tetrahedron Lett., 1968, 9, 6301–6304 CrossRef.
- J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14002–14007 CrossRef CAS.
- N. B. Perry, J. W. Blunt, M. H. G. Munro and L. K. Pannell, J. Am. Chem. Soc., 1988, 110, 4850–4851 CrossRef CAS.
- S. Sakemi, T. Ichiba, S. Kohmoto, G. Saucy and T. Higa, J. Am. Chem. Soc., 1988, 110, 4851–4853 CrossRef CAS.
- N. Fusetani, T. Sugawara and S. Matsunaga, J. Org. Chem., 1992, 57, 3828–3832 CrossRef CAS.
- J. Ahlert, E. Shepard, N. Lomovskaya, E. Zazopoulos, A. Staffa, B. O. Bachmann, K. X. Huang, L. Fonstein, A. Czisny, R. E. Whitwam, C. M. Farnet and J. S. Thorson, Science, 2002, 297, 1173–1176 CrossRef CAS.
- W. Liu, S. D. Christenson, S. Standage and B. Shen, Science, 2002, 297, 1170–1173 CrossRef CAS.
- N. Oku, S. Matsunaga and N. Fusetani, J. Am. Chem. Soc., 2003, 125, 2044–2045 CrossRef CAS.
- T. Nishizawa, M. Asayama, K. Fujii, K. Harada and M. Shirai, J. Biochem., 1999, 126, 520–529 CAS.
- G. Christiansen, J. Fastner, M. Erhard, T. Borner and E. Dittmann, J. Bacteriol., 2003, 185, 564–572 CrossRef CAS.
- C. E. Salomon, T. Deerinck, M. H. Ellisman and D. J. Faulkner, Mar. Biol., 2001, 139, 313–319 Search PubMed.
- J. D. Walton, Fung. Gen. Biol., 2000, 30, 167–171 Search PubMed.
- G. S. Sidhu, Eur. J. Plant Pathol., 2002, 108, 705–711 CrossRef CAS.
- I. Manzanares, C. Cuevas, R. Garcia-Nieto, E. Marco and F. Gago, Curr. Med. Chem. Anti-Cancer Agents, 2001, 1, 257–276 Search PubMed.
- C. Moss, D. H. Green, B. Perez, A. Velasco, R. Henriquez and J. D. McKenzie, Mar. Biol., 2003, 143, 99–110 Search PubMed.
- C. E. Salomon, L. E. Waggoner and M. G. Haygood, 2003, manuscript in progress.
- Y. Mikami, K. Takahashi, K. Yazawa, T. Arai, M. Namikoshi, S. Iwasaki and S. Okuda, J. Biol. Chem., 1985, 260, 344–348 CAS.
- R. G. Kerr and N. F. Miranda, J. Nat. Prod., 1995, 58, 1618–1621 CrossRef CAS.
- M. Kobayashi, S. J. Aoki, N. Ohyabu, M. Kurosu, W. Q. Wang and I. Kitagawa, Tetrahedron Lett., 1994, 35, 7969–7972 CAS.
- T. Golakoti, J. Ogino, C. E. Heltzel, T. Lehusebo, C. M. Jensen, L. K. Larsen, G. M. L. Patterson, R. E. Moore, S. L. Mooberry, T. H. Corbett and F. A. Valeriote, J. Am. Chem. Soc., 1995, 117, 12030–12049 CrossRef CAS.
- H. Shigemori, M. A. Bae, K. Yazawa, T. Sasaki and J. Kobayashi, J. Org. Chem., 1992, 57, 4317–4320 CrossRef CAS.
- S. P. Gunasekera, M. Gunasekera and P. McCarthy, J. Org. Chem., 1991, 56, 4830–4833 CrossRef CAS.
- S. Ito and Y. Hirata, Bull. Chem. Soc. Jpn., 1977, 50, 1813–1820 CAS.
- T. M. Zabriskie, J. A. Klocke, C. M. Ireland, A. H. Marcus, T. F. Molinski, D. J. Faulkner, C. F. Xu and J. C. Clardy, J. Am. Chem. Soc., 1986, 108, 3123–3124 CrossRef.
- R. Jansen, B. Kunze, H. Reichenbach and G. Hofle, Liebigs Ann. Chem., 1996, 285–290 Search PubMed.
- M. V. Dauria, L. G. Paloma, L. Minale, A. Zampella, J. F. Verbist, C. Roussakis and C. Debitus, Tetrahedron, 1993, 49, 8657–8664 CrossRef.
- M. Ishibashi, R. E. Moore, G. M. L. Patterson, C. F. Xu and J. Clardy, J. Org. Chem., 1986, 51, 5300–5306 CrossRef CAS.
- F. Sasse, H. Steinmetz, G. Hofle and H. Reichenbach, J. Antibiot., 1993, 46, 741–748 CAS.
- R. E. Schwartz, C. F. Hirsch, D. F. Sesin, J. E. Flor, M. Chartrain, R. E. Fromtling, G. H. Harris, M. J. Salvatore, J. M. Liesch and K. Yudin, J. Ind. Microbiol., 1990, 5, 113–123 Search PubMed.
- N. A. Margarvey and C. E. Salomon, unpublished data.
- N. Magarvey, J. Keller, V. Bernan, M. Dworkin, D. H. Sherman, submitted to Appl. Environ. Microbiol., 2003 Search PubMed.
- S. K. Davidson and M. G. Haygood, Biol. Bull., 1999, 196, 273–280 Search PubMed.
- S. P. Gunasekera, M. Gunasekera, R. E. Longley and G. K. Schulte, J. Org. Chem., 1990, 55, 4912–4915 CrossRef CAS.
- D. T. Hung, J. Chen and S. L. Schreiber, Chem. Biol., 1996, 3, 287–293 CrossRef CAS.
- R. J. Kowalski, P. Giannakakou, S. P. Gunasekera, R. E. Longley, B. W. Day and E. Hamel, Mol. Pharm., 1997, 52, 613–622 Search PubMed.
- E. terHaar, R. J. Kowalski, E. Hamel, C. M. Lin, R. E. Longley, S. P. Gunasekera, H. S. Rosenkranz and B. W. Day, Biochemistry, 1996, 35, 243–250 CrossRef CAS.
- I. Paterson and G. J. Florence, Eur. J. Org. Chem., 2003, 2193–2208 CrossRef CAS.
- G. Imsiecke, M. Custodio, R. Borojevic, R. Steffen, M. A. Moustafa and W. E. G. Muller, Cell Biol. Int., 1995, 19, 995–1000 CrossRef CAS.
- M. J. Garson, A. E. Flowers, R. I. Webb, R. D. Charan and E. J. McCaffrey, Cell Tissue Res., 1998, 293, 365–373 CrossRef CAS.
- (a) H. Xu, Z. X. Wang, J. Schmidt, L. Heide and S. M. Li, Mol. Genetics Genomics, 2002, 268, 387–396 Search PubMed; (b) L. Du, C. Sanchez, M. Chen, D. Edwards and B. Shen, Chem. Biol., 2000, 7, 623–642 CrossRef CAS; (c) J. J. Coque, F. J. Perez-Llarena, F. J. Enguita, J. L. Fuente, J. F. Martin and P. Liras, Gene, 1995, 162, 21–27 CrossRef CAS; (d) Y. Mao, M. Varoglu and D. H. Sherman, Chem. Biol., 1999, 6, 251–263 CrossRef CAS.
- K. L. Rinehart, Med. Res. Rev., 2000, 20, 1–27 CrossRef CAS.
- M. D. Vera and M. M. Joullie, Med. Res. Rev., 2002, 22, 102–145 CrossRef CAS.
- G. L. Challis, J. Ravel and C. A. Townsend, Chem. Biol., 2000, 7, 211–224 CrossRef CAS.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS.
- M. G. Haygood and S. K. Davidson, Appl. Environ. Microbiol., 1997, 63, 4612–4616 CAS.
- N. S. Webster, K. J. Wilson, L. L. Blackall and R. T. Hill, Appl. Environ. Microbiol., 2001, 67, 434–444 CrossRef CAS.
- S. A. Waksman and H. B. Woodruff, Proc. Soc. Exp. Biol. Med., 1940, 45, 609–614 Search PubMed.
- H. W. Florey, E. Chain, N. G. Heatley, M. A. Jennings, A. G. Sanders, E. P. Abraham and M. E. Florey, Antibiotics: a survey of penicillin, streptomycin, and other antimicrobial substances from fungi, actinomycetes, bacteria, and plants, Oxford University Press: City, 1949 Search PubMed.
- N. S. Webster, K. J. Wilson, L. L. Blackall and R. T. Hill, Appl. Environ. Microbiol., 2001, 67, 434–444 CrossRef CAS.
- U. Hentschel, M. Schmid, M. Wagner, L. Fieseler, C. Gernert and J. Hacker, FEMS Microbiol. Ecol., 2001, 35, 305–312 CrossRef CAS.
- A. A. Stierle, J. H. Cardellina and F. L. Singleton, Tetrahedron Lett., 1991, 32, 4847–4848 CrossRef CAS.
- Z. H. Zheng, W. Zeng, Y. J. Huang, Z. Y. Yang, J. Li, H. R. Cai and W. J. Su, FEMS Microbiol. Lett., 2000, 188, 87–91 CrossRef CAS.
- R. H. Feling, G. O. Buchanan, T. J. Mincer, C. A. Kauffman, P. R. Jensen and W. Fenical, Angew. Chem., Int. Ed., 2003, 42, 355–357 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |