Complexation behaviour of hexadentate ligands possessing N2O4 and N2O2S2 cores: differential reactivity towards Co(II), Ni(II) and Zn(II) salts and structures of the products
Gudneppanavar
Rajsekhar
a,
Chebrolu P.
Rao
*a,
Pauli
Saarenketo
b,
Kalle
Nättinen
b and
Kari
Rissanen
b
aBioinorganic Laboratory, Department of Chemistry, Indian Institute of Technology Bombay, Mumbai, 400 076, India. E-mail: cprao@chem.iitb.ac.in
bDepartment of Chemistry, University of Jyvaskyla, Jyvaskyla, SF - 40351, Finland
First published on 30th October 2003
Abstract
Reactions of divalent metal salts of Co, Ni and Zn with 1,2-di(salicylaldimino-o-phenylthio)ethane (H2L1) and 1,2-di(naphthaldimino-o-phenylthio)ethane (H2L2), having N2O2S2 cores, and 1,2-di(O-salicylaldimino-o-hydroxyphenyl)ethane (H2L3), having a N2O4 core, have been explored. Out of the three ligands and the nine products obtained from the corresponding reactions, two ligands and seven products were crystallographically characterized. However, all the ligands and the products were characterized by analytical and spectral methods. Reaction of H2L1 and H2L2 with Co(II) salts results in oxidative cleavage of the C–S bond to produce a Co(III) product bound to two dissimilar tridentate ligands formed as a result of the cleavage. The reaction of H2L1 and H2L2 with Co(III)acac in the presence of methanolic NaOH also shows cleavage of the C–S bond to give compounds similar to those obtained by reacting H2L1 and H2L2 with Co(II) salts in the absence of additional external base. However, the reaction of Co(II) salt with H2L3 did not lead to any C–O bond cleavage; rather it produced an octahedral Co(II) complex of the hexadentate H2L3. Reactions of all these three ligands with Ni(II) salt resulted in octahedral complexes of the corresponding hexadentate ligands. In the case of Zn(II), while H2L3 with a N2O4 core resulted in an octahedral complex, H2L1 and H2L2, both with a N2O2S2 core, produced pseudo-octahedral complexes whose Zn–S bond lengths are rather long. The conformations of both the 5-membered and 6-membered chelate rings formed in the products were evaluated. The extent of distortion exhibited in the geometry of these octahedral complexes was computed and appropriately compared.
Introduction
Molecules possessing O, N and S donor sites are important in the development of coordination chemistry as well as in the bio-mimetic chemistry of a number of metal ions, particularly the transition metals. Among these, non-cyclic hexadentate ligands with N2O4 and N2O2S2 binding cores are well-suited for the coordination of transition metal ions with a high preference towards the later ones.1 A non-cyclic ligand of the N2O2S2 type, 1,2-di(salicylaldimino-o-phenylthio)ethane (H2L1), generated interesting chemistry with oxidative cleavage of the ligand when it was reacted with a Co(II) salt, rather than forming a simple complex.2 However, an earlier work reported the formation of only a simple cobalt complex from the same reaction but the product was not subjected to NMR or X-ray studies.3 C–S bond cleavage is not an unusual phenomenon, particularly with cobalt group elements in the presence of cyclic thioether ligands as well as with some non-cyclic ones,4 but this is not the case for the ligands in this paper. In the literature,4 the suitable size for the cleavage of thia-chelates was addressed and in such cases the C–H bond is activated by a base, such as Et3N or NaOH/KOH. The present study differs even in this context. All these points aroused great interest in our group to understand what happens to the cleavage reaction when (a) the sulfur is replaced by oxygen, (b) Co(II) is replaced by Ni(II) or Zn(II), (c) the counter ion of the Co(II) salt is varied and (d) Co(II) is replaced by a Co(III) salt. These aspects were addressed in a systematic manner by developing three types of hexadentate ligands (H2L1, H2L2 and H2L3), having either an N2O4 core or an N2O2S2 core, and studying their reactivity and coordination chemistry using three divalent metal ions, viz., Co(II), Ni(II) and Zn(II) and four different Co(II) salts as well as one acetylacetonate (acac) compound of Co(III). In the process, the reaction product in each case was isolated, purified and characterized. Structures of several of these products, including the precursor ligands, were established by single crystal X-ray diffraction (XRD). The results of all these studies are reported in this paper in a comparative way.Results and discussion
All the compounds reported in Scheme 1, except those of Ni(II) (2, 5 and 8), were characterized by 1H and 13C NMR and all were characterized by FAB-MS, UV-Vis and magnetic moment measurements. Except for H2L2, 4 and 2, all other ligands and complexes were structurally characterized by single crystal XRD.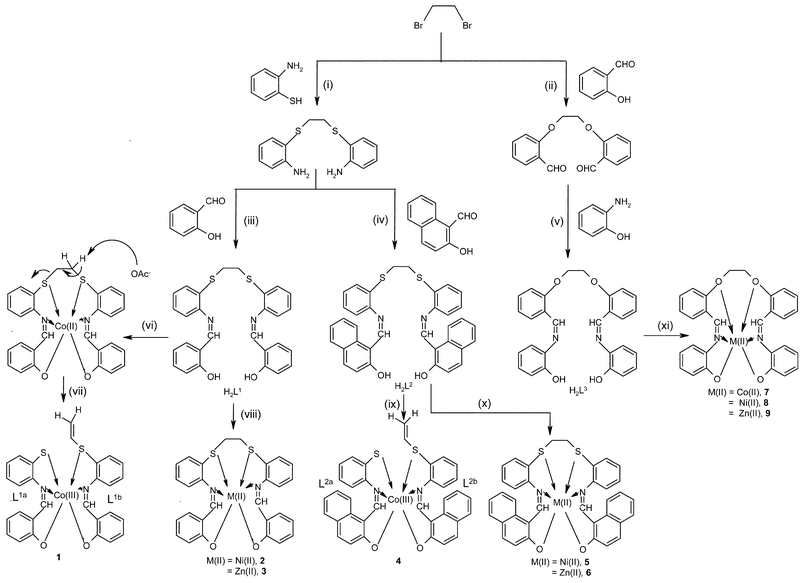 | ||
| Scheme 1 Schematic representation of the synthesis of the ligands and their Co(III), Co(II), Ni(II) and Zn(II) complexes. (i) Dry ethanol and Na; (ii) aqueous NaOH; (iii) MeOH, RT, stirring; (iv) MeOH, RT, stirring; (v) EtOH, reflux for 6 h; (vi) Co(OAc)2·4H2O, argon/Co(acac)3/NaOH; (vii) oxygen; (viii) Ni(OAc)2·4H2O/Zn(OAc)2·2H2O; (ix) Co(OAc)2·4H2O, argon, oxygen/Co(acac)3, NaOH; (x) Ni(OAc)2·4H2O/Zn(OAc)2·2H2O; (xi) Co(OAc)2·4H2O/Ni(OAc)2·4H2O/Zn(OAc)2·2H2O. | ||
1. Reactions of H2L1–H2L3 with Co(II)
While the reaction of Co(II) salts (acetate, chloride, sulfate and acetylacetonate) with H2L1 or H2L2 having an N2O2S2 core results in the oxidative cleavage of the C–S bond, the reaction of H2L3 having an N2O4 core produces only a simple Co(II) complex.The reactions were initially allowed to proceed under Ar atmosphere and later on under O2. During this process, the reaction mixture goes through a color change from orange-red to dark brown. The reaction of H2L1 with Co(acetate)2·4H2O was monitored in solution by UV-Vis absorption spectra as a function of time. It was noticed that when the reaction mixture was maintained under Ar atmosphere, the orange-red complex remains stable, but when the reaction was purged with O2, it quickly converted to a dark brown Co(III) complex with cleavage of H2L1 as shown in Scheme 1. Thus, under Ar atmosphere the reaction mixture exhibited a ligand![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) →metal charge transfer band at 425 nm in the absorption spectra and this band was found to grow in intensity as a function of time; this peak is reminiscent of a Co(II) center. When the reaction mixture was bubbled with O2, the spectra exhibited a very broad band, which upon de-convolution represents two least resolved bands positioned at 440 and 475 nm, respectively, with almost equal absorbance, indicating the formation of a more positive Co(III) center, favoring a better charge transfer from the thiolate moiety that was generated by cleavage (L1a). Similarly, the band observed at 300 nm under Ar atmosphere shifts to 285 nm when purged with O2. Even the absorption spectrum measured from the dichloromethane solution of the isolated product exhibited a spectrum that is exactly identical to that obtained from the reaction mixture after O2 purging. Similarly, in the case of 4 the absorption spectrum measured from the isolated product exhibited a broad band, which upon de-convolution represents two less resolved bands positioned at 480 and 510 nm.
→metal charge transfer band at 425 nm in the absorption spectra and this band was found to grow in intensity as a function of time; this peak is reminiscent of a Co(II) center. When the reaction mixture was bubbled with O2, the spectra exhibited a very broad band, which upon de-convolution represents two least resolved bands positioned at 440 and 475 nm, respectively, with almost equal absorbance, indicating the formation of a more positive Co(III) center, favoring a better charge transfer from the thiolate moiety that was generated by cleavage (L1a). Similarly, the band observed at 300 nm under Ar atmosphere shifts to 285 nm when purged with O2. Even the absorption spectrum measured from the dichloromethane solution of the isolated product exhibited a spectrum that is exactly identical to that obtained from the reaction mixture after O2 purging. Similarly, in the case of 4 the absorption spectrum measured from the isolated product exhibited a broad band, which upon de-convolution represents two less resolved bands positioned at 480 and 510 nm.
From the FTIR spectra of 1 and 4, it was possible to deduce the binding of the –O−phenolate group owing to the disappearance of the corresponding νOH vibration that is otherwise present in the precursor ligand spectra. Involvement of the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– moiety in binding the metal ion center is evident based on the shift (8–20 cm−1) observed in the stretching frequency of this group in the complexes. Both the complexes exhibited a strong νCC vibration arising from the –S–HCCH2 moiety formed as a result of the C–S bond cleavage in L1 and L2 at 1525 and 1532 cm−1, respectively. The molecular weights of the ligands as well as the complexes were confirmed based on the molecular ion peaks observed in their respective FAB mass spectra. The results are consistent with mononuclear complexes. Both the complexes were found to be diamagnetic from their magnetic susceptibility measurements, supporting the +3 oxidation state of the cobalt.
N– moiety in binding the metal ion center is evident based on the shift (8–20 cm−1) observed in the stretching frequency of this group in the complexes. Both the complexes exhibited a strong νCC vibration arising from the –S–HCCH2 moiety formed as a result of the C–S bond cleavage in L1 and L2 at 1525 and 1532 cm−1, respectively. The molecular weights of the ligands as well as the complexes were confirmed based on the molecular ion peaks observed in their respective FAB mass spectra. The results are consistent with mononuclear complexes. Both the complexes were found to be diamagnetic from their magnetic susceptibility measurements, supporting the +3 oxidation state of the cobalt.
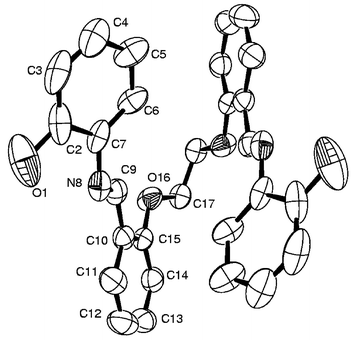
Molecular structure of H2L1. H2L1crystallizes in the monoclinic system with P21/c space group. The molecule sits on a crystallographic center of symmetry and hence each asymmetric unit cell possesses two halves of the molecule. The molecule exhibits an ‘S’-like structure. Such a molecular structure is stabilized via the presence of a strong intramolecular 6-atom O–H⋯N type interaction between the phenolic–O–H and the –N
C– moieties and another weak intramolecular 9-atom O–H⋯S type interaction between the phenolic–O–H and the Ph–S–CH2– moieties.
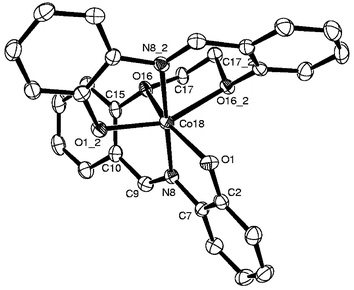
Crystal structure of 1 and the role of the anion of the Co(II) salt. The crystal structure of the reaction product, [Co(III)(L1a)(L1b)] (1) shown in Fig. 1 reveals the presence of two ligands, viz., L1a (dianionic) and L1b (monoanionic), formed upon cleavage of a C–S bond, both acting as tridentate ligands by extending the ONS binding core. This results in the formation of two 5-membered and two 6-membered chelate rings to give a distorted octahedral Co(ONS)2 core. The conformations of the various chelates present in the complex are shown in the Table 1. The octahedral coordination geometry is much less distorted compared with the regular octahedron geometry as shown by the observed narrower bite angles, 88.3(1)°–95.8(1)°, and the trans angles: 175.6(1)°, 175.6(1)° and 177.9(2)°. Selected bond lengths are given in Table 2 and the bond angles in Table 3. The metric data is fully supported by the cleavage of H2L1 in order to form the ethylene moiety C17b
C18b (1.302 Å) and Co–Sthiolate
(2.227 Å) and Co–Sthioether
(2.238 Å) bonds. No abnormal bond lengths were observed in the structure.
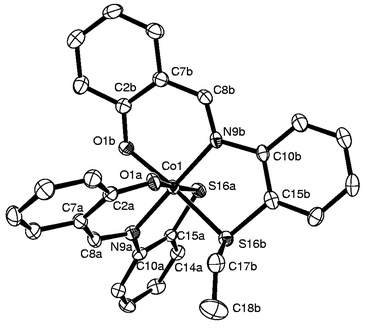 | ||
| Fig. 1 Molecular structure with atom labeling for 1. | ||
| Complex | Chelate | Conformationa |
|---|---|---|
|
a
C
2=half-chair form; Cs=envelop form; A=planar
|
||
| 1 | Co1–N9a–C10a–C15a–S16a | C s |
| Co1–S16b–C15b–C10b–N9b | C 2 | |
| Co1–O1b–C2b–C7b–C8b–N9b | Skew boat | |
| Co1–O1a–C2a–C7a–C8a–N9a | Boat | |
| 7 | Co18–N8–C7–C2–O1 | C s |
| Co18–O16–C17–C17_2–O16_2 | A | |
| Co18–N8–C9–C10–C15–O16 | Skew boat | |
| 5 | Ni1–S23–C22–C21–S20 | C 2 |
| Ni1–S20–C19–C14–N13 | C s | |
| Ni1–S23–C24–C29–N30 | C 2 | |
| Ni1–N30–C31–C32–C41–O42 | Skew boat | |
| Ni1–N13–C12–C11–C2–O1 | Boat | |
| 8 | Ni18–O1–C2–C7–N8 | C s |
| Ni18–O16–C17–C17_2–O16_2 | A | |
| Ni18–N8–C9–C10–C15–O16 | Skew boat | |
| 3 | Zn1–S16–C17–C18–S19 | C 2 |
| Zn1–S16–C15–C10–N9 | C 2 | |
| Zn1–S19–C20–C25–N26 | C s | |
| Zn1–N26–C27–C28–C33–O34 | Skew boat | |
| Zn1–N9–C8–C7–C2–O1 | Skew boat | |
| 6 | Zn1–S20–C21–C21_2–S20_2 | A |
| Zn1–S20–C19–C14–N13 | C s | |
| Zn1–N13–C12–C11–C2–O1 | Skew boat | |
| 9 | Zn18–O1–C2–C7–N8 | C 2 |
| Zn18–N8–C9–C10–C15–O16 | Skew boat | |
| Zn18–O16–C17–C17_2–O16_2 | A | |
| Bond | [Co(III)(L1a)(L1b)] (1) | [Co(L3)]·CH3COOH (7)a | [Ni(L2)]·0.5CH3OH (5) | [Ni(L3)]·CH3COOH (8)a | [Zn(L1)] (3) | [Zn(L2)] (6)a | [Zn(L3)]·CH3OH (9)a |
|---|---|---|---|---|---|---|---|
| a Molecule possessing a center of symmetry. | |||||||
| M–N | 1.913(3) | 2.066(2) | 2.028(3) | 2.018(4) | 2.095(3) | 2.073(2) | 2.151(2) |
| 1.926(2) | 2.028(3) | 2.101(4) | |||||
| M–Ophenolate | 1.894(2) | 2.007(2) | 2.008(3) | 2.010(4) | 1.980(3) | 1.978(2) | 2.020(2) |
| 1.914(2) | 2.014(3) | 2.009(3) | |||||
| M–Oether | – | 2.217(2) | – | 2.178(3) | – | – | 2.328(2) |
| M–Sether | 2.227(1) | – | 2.413(2) | –- | 2.582(2) | 2.692(1) | – |
| 2.238(1) | 2.459(2) | 2.675(2) | |||||
| [Co(III)(L1a)(L1b)] (1) | [Co(L3)]·CH3COOH (7) | [Ni(L2)]·0.5CH3OH (5) | [Ni(L3)]·CH3COOH (8) | [Zn(L1)] (3) | [Zn(L2)] (6) | [Zn(L3)]·CH3OH (9) | |
|---|---|---|---|---|---|---|---|
| a Co(III) bound to thioether. b Co(III) bound to thiolate. | |||||||
| Ophe–M–Ophe. | 87.89(9) | 112.53(12) | 93.67(12) | 106.0(2) | 104.25(13) | 103.01(11) | 111.57(11) |
| Ophe–M–N | 84.93(10) | 81.99(9) | 88.45(13) | 96.11(16) | 91.82(12) | 88.33(8) | 82.75(8) |
| 95.81(10) | 98.26(9) | 90.28(13) | 82.78(16) | 99.45(13) | 100.22(8) | 104.07(8) | |
| 95.23(10) | 89.49(13) | 96.68(12) | |||||
| 86.25(10) | 88.33(13) | 90.07(12) | |||||
| Ophe–M–S | 175.64(7)a | – | 90.38(10) | – | 168.21(10) | 162.82(5) | – |
| 92.28(7)a | 170.90(9) | 86.03(9) | 89.35(6) | ||||
| 94.07(7)b | 170.62(9) | 90.50(9) | |||||
| 175.64(7)b | 90.23(8) | 162.64(9) | |||||
| Ophe–M–Oether | – | 155.66(8) | – | 161.37(14) | – | – | 88.75(8) |
| 88.30(8) | 89.29(14) | 156.60(7) | |||||
| N–M–S | 90.72(8)a | – | 97.98(11) | – | 80.66(10) | 77.63(6) | – |
| 89.13(8)b | 83.55(11) | 88.94(10) | 91.94(6) | ||||
| 88.25(8)b | 82.99(9) | 89.01(10) | |||||
| 89.69(8)b | 99.17(10) | 78.99(10) | |||||
| N–M–Oether | – | 82.65(9) | – | 96.41(15) | – | – | 89.43(8) |
| 96.99(9) | 85.02(15) | 81.04(8) | |||||
| N–M–N | 177.94(12) | 179.54(14) | 177.44(14) | 178.2(2) | 165.31(13) | 166.33(11) | 168.06(12) |
| S–M–S | 86.07(3) | – | 87.02(4) | – | 80.38(4) | 81.38(3) | – |
| Oether–M–Oether | – | 74.96(10) | – | 78.12(18) | – | – | 74.35(10) |
A similar reaction carried out with another salt of Co(II), CoCl2·6H2O, and H2L1 resulted in a product whose composition and crystal structure were found to be the same as that of [Co(III)(L1a)(L1b)]. Based on FAB mass and 1H NMR studies, it was found that the reaction carried out using Co(SO4)2·7H2O and Co(acac)2·2H2O also resulted in the same product and thereby exhibited C–S bond cleavage. Thus, in these four cases the final product was found to be same, irrespective of the anion of the Co(II) salt used.
1H and 13C NMR studies. The absence of paramagnetic shifts in the 1H NMR spectrum of 4 supports the presence of a Co(III) center formed by the oxidation of Co(II). XRD supports the Co being present in its 3+ state as well as the presence of the C–S bond-cleaved fragments (L1a and L1b) bound to the metal ion center as reported in this paper. This was further supported by the 1H NMR spectrum of 1. The spectrum of 4 also clearly supports the formation of two ligands, L2a and L2b, formed by the cleavage of a C–S bond in the reaction between Co(II) acetate and H2L2: the imine proton peaks are doubled and new signals from vinylic protons arise, as shown in Fig. 2(a). The same information is also revealed through the spectral changes observed in case of the aromatic protons. These features are vividly highlighted when the spectrum of 4 is compared with that of the precursor ligand, H2L2, given in Fig. 2(b). The peak observed at 3.195 ppm due to the bridging –S–CH2–CH2–S– moiety in H2L2 disappears in the spectrum of 4 and two new multiplets appear in the region of 5.574–5.957 ppm, which are due to the vinyl protons (–S–CH
CH2), the assignment of which is made using 1H–1H COSY as shown by the cross peak in Fig. 3(a). Based on the 1H–13C HMQC experiments [Fig. 3(b)], the 13C peaks observed at 128.8 and 126.9 ppm were assigned to S–CH=CH2 and –S–CHCH2, respectively. The peak doubling for imine protons was further confirmed through the 1H–13C HMQC experiment shown in Fig. 3(a).
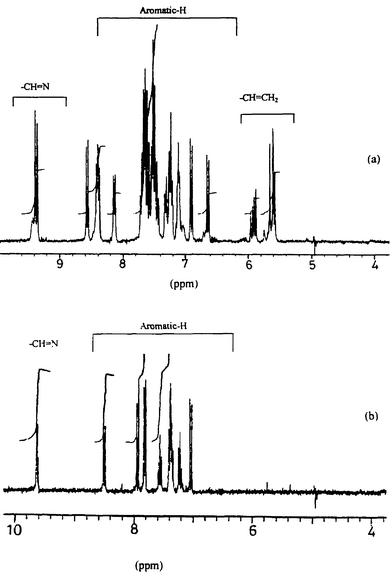
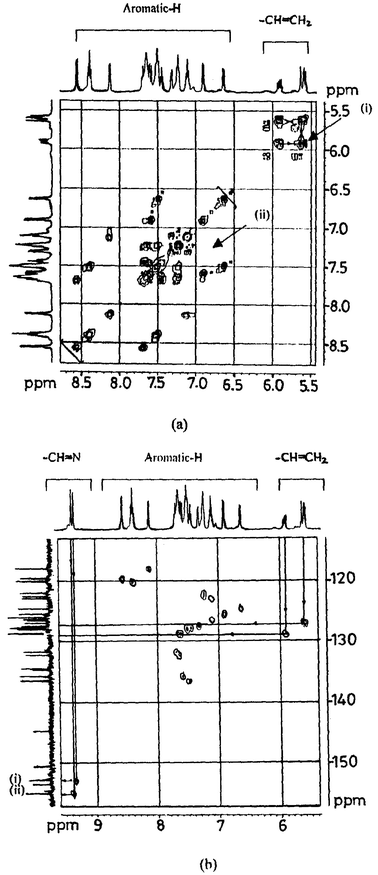 N was shifted from 1619 cm−1 in the ligand to 1598 cm−1 in the complex, indicating the binding of cobalt through –O− and CHN, respectively. The spectrum of crystals of 7 exhibited νCO at 1700 cm−1 and two sharp peaks for νCH at 2924 and 2853 cm−1. When these crystals were crushed, washed with diethyl ether and dried in vacuo, the peaks at 1700, 2924 and 2853 cm−1 disappeared, indicating the loss of acetic acid otherwise present in the lattice. The molecular weight of H2L3 and [Co(L3)] were confirmed based on their FAB mass spectra. The observed magnetic moment value for 7
(μobs=4.899 BM) arises from the cobalt in its +2 oxidation state along with orbital contribution expected to arise from a high-spin d7 system. In the 1H NMR spectrum of the ligand, H2L3, only the peaks arising from one-half of the molecule were observed owing to the symmetry. On the other hand, the 1H NMR spectrum of 7 exhibited paramagnetically shifted bands in the region −25 to +25 ppm.
N was shifted from 1619 cm−1 in the ligand to 1598 cm−1 in the complex, indicating the binding of cobalt through –O− and CHN, respectively. The spectrum of crystals of 7 exhibited νCO at 1700 cm−1 and two sharp peaks for νCH at 2924 and 2853 cm−1. When these crystals were crushed, washed with diethyl ether and dried in vacuo, the peaks at 1700, 2924 and 2853 cm−1 disappeared, indicating the loss of acetic acid otherwise present in the lattice. The molecular weight of H2L3 and [Co(L3)] were confirmed based on their FAB mass spectra. The observed magnetic moment value for 7
(μobs=4.899 BM) arises from the cobalt in its +2 oxidation state along with orbital contribution expected to arise from a high-spin d7 system. In the 1H NMR spectrum of the ligand, H2L3, only the peaks arising from one-half of the molecule were observed owing to the symmetry. On the other hand, the 1H NMR spectrum of 7 exhibited paramagnetically shifted bands in the region −25 to +25 ppm.
Molecular structure of H2L3 and 7. Both the structures of H2L3 and 7 possess a center of symmetry and have only one-half of the molecule in their asymmetric unit cells. The ORTEP plots of H2L3 and 7 are shown in Figs. 4 and 5, respectively. The molecule of H2L3 is folded due to one C–H⋯O (5-atom) type intramolecular hydrogen bond interaction present between the –CH
N– and the Ph–O–CH2– moieties and one O–H⋯N (5-atom) type interaction present between the phenolic–O–H and the –CHN– moieties. In the structure of 7, the cobalt is present in a distorted octahedral geometry bound through two phenolic moieties, two imine moieties and two ether oxygens, resulting in a neutral mononuclear complex with a Co(II)–N2O4 core. Thus, the X-ray structure clearly shows an intact C–O bond and supports the specificity of Co(II) salts towards the cleavage of C–S bonds rather than the C–O bond. Among the coordinating atoms the imine nitrogens occupy axial positions and the remaining four oxygen atoms occupy the equatorial positions. The octahedral geometry is more distorted than the regular octahedron geometry as shown by their wider range of bite angles, 75.0(1)° to 112.5(2)°, and trans angles, 155.7(1)° and 179.5(2)°. The conformations of the chelates found in 7 are given in Table 1. Selected bond lengths and bond angles are given in Tables 2 and 3, respectively. However, no abnormalities were found with the bond length data of H2L3 and 7. The M–N and M–Ophenolate distances observed in 7 with Co(II) are considerably longer, by about 0.15 and 0.10 Å, respectively, when compared with similar bond lengths observed in the structure of 1 with Co(III), due to the difference in the oxidation states.
 | ||
| Fig. 4 ORTEP drawing for the molecular structure of H2L3. The molecule sits on a center of symmetry. | ||
 | ||
| Fig. 5 Molecular structure for 7 as an ORTEP plot. Co(II) sits on a center of symmetry. | ||
2. Reactions of H2L1 and H2L2 with Co(III)
Reactions of H2L1 and H2L2 with Co(acac)3 in 1 ∶ 1 ratio at room temperature as well as under reflux conditions in dichloromethane–methanol did not yield any products. But the same reactions, when carried out in the presence of 2 equiv. of methanolic NaOH, resulted in the formation of 1 and 4, respectively, as confirmed by 1H NMR studies. Thus, the alkali activates the thioether group while deprotonating the phenolic –OH groups. This observation is in agreement with that reported in the literature.4i3. Reaction of H2L1–H2L3 with Ni(II)
Reactions of H2L1–H2L3 with Ni(CH3COO)2·4H2O resulted in the formation of the hexacoordinated Ni(II) complexes 2, 5 and 8, respectively, as shown in Scheme 1. In none of these final products were either the C–S or the C–O bonds broken upon reaction with Ni(II) salt, though C–S bond cleavage was reported in the literature in the case of a thioether complex of nickel.4j Thus, Ni(II) behaves quite differently from Co(II), where the latter is stabilized in the +3 oxidation state when cleaving the C–S bond with H2L1 and H2L2, while with H2L3 the final product is in the same +2 oxidation state and the C–O bond is intact. In the FTIR spectra of 2, 5 and 8 the observed decrease in νCHN
(8–19 cm−1) and the disappearance of νOH vibrations support the binding through –O− and –CHN– moieties. The analytical and FAB mass data fits with the mononuclear complexes shown in Scheme 1. The experimental magnetic moments were found to be 2.841, 3.049 and 2.812 BM, respectively, for 2, 5 and 8 at room temperature. The 1H NMR spectra of these complexes exhibited broad and shifted bands due to their paramagnetic nature, as supported by the magnetic moments. In the absorption spectra of the Ni(II) complexes 2, 5 and 8, bands corresponding to 3A2g→3T1g(F) and 3A2g→3T1g(P) transitions are observed at 567–624 and 445–471 nm, respectively, and a third band due to 3A2g→3T2g at 879–908 nm supports the octahedral geometry around the Ni(II) center.
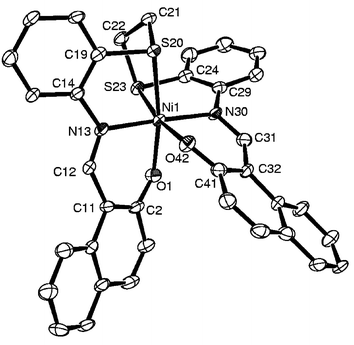 | ||
| Fig. 6 Molecular structure for 5 as an ORTEP plot. | ||
4. Reactions of H2L1–H2L3 with Zn(II)
The products 3, 6 and 9 are obtained by reacting Zn(CH3COO)2·2H2O with H2L1, H2L2 and H2L3, respectively. The product formulations were supported by the observed satisfactory elemental analysis data as well as the molecular weight obtained through the molecular ion peak in the FAB mass spectra. For these diamagnetic Zn(II) complexes, the 1H NMR spectra exhibited peaks corresponding to half of the ligand, suggesting retention of the ligand symmetry even in the bound state. The 1H NMR peaks arising from the –S–CH2–CH2–S– moiety in the case of 3 and 6 are found to be overlapped by the broad peak arising from water in DMSO-d6. On the other hand the peak arising from –O–CH2–CH2–O– in 9 was observed at 4.384 ppm. Disappearance of the peak due to phenolic –OH and an upfield shift observed in the peak for the –CHN proton both indicate that the Zn(II) is bound through the azomethine and phenolic groups in 3, 6 and 9.
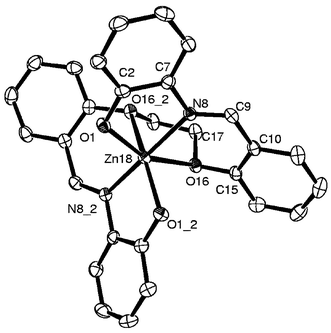 | ||
| Fig. 7 Molecular structure for 9 as an ORTEP plot. Zn(II) sits on a center of symmetry. | ||
The bond lengths observed in 9, Zn–Oether=2.328(2), Zn–Ophenolic=2.020(2) and Zn–N=2.151(2)
Å, are quite normal when compared with the corresponding distances reported in the literature and the metric parameters fit well with an octahedral geometry around the Zn(II) center. The observed Zn–Oether distances in the present case are well within the range reported for a Zn(II)-crown ether complex (2.34–2.60 Å).5a Recently, we reported a weak interaction of the pyranose oxygen with the Zn(II) center, resulting in a Zn–O distance of 2.759 Å.5b
On the other hand, in the case of 3 and 6 the bond lengths, Zn–Ophenolic=1.978(2)–2.009(1)
Å and Zn–N=2.073(1)–2.101(1)
Å, are well within the covalent bond distances for the corresponding pairs, while the Zn–S distances are in the range of 2.582(1)–2.692(1)
Å, well within the sum of the van der Waals radii. In the literature, the Zn–S distances in thiolates are observed to be around 2.300–2.360 Å for terminal bonds and 2.400–2.450 Å for bridging ones.5c The Zn–S distances observed in the present case are somewhat longer than that observed in the crown thioether (2.497 Å) and shorter than that observed in the complex of dithiaalkyl-substituted triazine 1-oxide (2.649–2.720 Å).5d,1c The Zn(II) exhibits pseudo-octahedral geometry. However, when these two Zn–S interactions are disregarded the resulting ZnN2O2 core does not fit well with a tetrahedral geometry, strongly suggesting that the coordination sphere is more of a distorted octahedral rather than tetrahedral. The selected bond lengths of the coordination sphere are given in Table 2.
Conclusions and comparisons
The complexes of Co(III), Co(II), Ni(II) and Zn(II) with H2L1–H2L3 reported in this paper are all neutral and found to have distorted octahedral geometries, however, 3 and 6 have a pseudo-octahedral geometry. The present study clearly demonstrates the formation of two tridentate ligands from the precursor hexadentate ligands (N2O2S2, H2L1 and H2L2) via C–S bond cleavage as a result of the oxidation of Co(II). Such C–S bond cleavage was found to occur with the Co(II) salts of chloride, sulfate, acetate and acetylacetonate. On the other hand, H2L3, possessing a N2O4 core, does not exhibit cleavage of the C–O bond under the same conditions. On going from Co(III) (1) to the Co(II) (7) complex, the corresponding bond lengths in the primary coordination sphere are considerably increased, consistent with the change in the oxidation state of cobalt.To observe the role of the metal ion, we extended these studies using Ni(II) and Zn(II) salts. Combining the present studies with what is known in the literature, the following aspects are relevant for bond cleavage: (a) the presence of ‘S’ instead of ‘O’, (b) the metal ion should support various oxidation states and (c) the thioether chelate should be 5-membered.
In all the complexes the two imine nitrogens are trans- to each other. On going from a trivalent complex (1, Co3+) to divalent complexes (Co2+: 7; Ni2+: 5 and 8; Zn2+: 3, 6 and 9) the primary coordination distances, viz., M–N, M–Ophenolate or M–S, generally increase by 0.1 Å or more owing to the change in size of the corresponding metal ion center. These distances increase further in the case of the Zn2+ complexes as compared to the Ni2+ or Co2+ complexes, owing to expansion of the coordination sphere of Zn2+ from its commonly observed tetrahedral geometry to the pseudo-octahedral one.
The Co(II), Ni(II) and Zn(II) complexes derived from H2L3 are more distorted than the Co(III), Ni(II) and Zn(II) complexes derived from the other two ligands, viz., H2L1 and H2L2. The extent of distortion observed about the metal ion coordination sphere in these complexes follows a trend, Zn(II)>Co(II)>Ni(II), as can be deduced from the corresponding bond lengths and bond angles. This can also be seen from the stereo views of the complexes 7, 8 and 9 given in Fig. 8. Among all the complexes, 1 with Co(III) has the least distortion due to cleavage of the hexadentate ligand into two tridentate ligands.
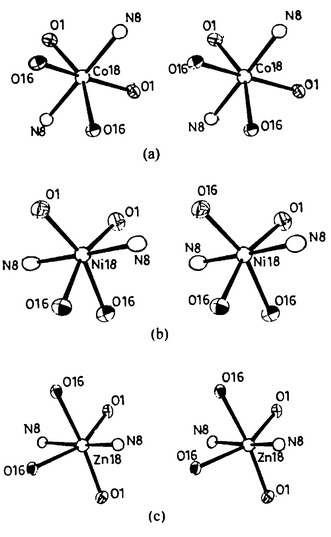 | ||
| Fig. 8 Stereo views of the primary coordination spheres for the complexes (a) 7 (CoN2O4), (b) 8 (NiN2O4) and (c) 9 (ZnN2O4). | ||
The conformation of the 5-membered rings can be half chair (C2), envelope (Cs) or planar (A), while the six-membered rings exhibit boat or skew boat conformations.
Experimental details
General remarks
1,2-Dibromoethane was procured from Spectrochem Pvt. Ltd.; o-hydroxynapthaldehyde and 1,2-di(2-aminophenylthio)ethane were synthesized as per the reported procedures.6a,6b Ethylene-2-2′-(dioxydibenzaldehyde) was synthesized as per the reported procedure but with some modifications.6c All the solvents were purified and dried immediately before use. Elemental analysis was carried out on a CE Instruments Flash EA 1112 series apparatus; FTIR spectra were recorded on Nicolet Magna IR 550; UV-Vis spectra were recorded on a UV-2101PC spectrophotometer. FAB mass spectra were recorded on a JEOL SX 102/DA-6000 mass spectrometer data system using argon–xenon (6 kV, 10 mA) as the FAB gas. The accelerating voltage was 10 kV and the spectra were recorded at room temperature with m-nitrobenzyl alcohol (NBA) as the matrix. GC-mass spectra were recorded on a 5989 B MS engine model from Hewlett Packard. 1H NMR spectra were recorded on a Varian VXR 300S. 13C spectra were recorded on a Bruker Avance DRX 500 spectrometer.Syntheses
×40 ml), then combined with the original organic phase. The organic layer was then washed with 10% sodium hydroxide (20 ml) and dried over MgSO4. The dichloromethane was removed by rotary evaporation and the resulting residue was cooled at 4°C overnight. The solid thus separated was filtered and was washed with diethyl ether and then dried under vacuum. The colorless solid was further crystallized from acetone or hot petroleum ether (5.5 g, 16%). M.p. 110–112°C. 1H-NMR (300 MHz, CDCl3)
δ: 4.520 (s, 4H, –OCH2CH2O–), 7.031–7.842 (m, 8H, aromatic H), 10.412 (s, 2H, –CHO). FTIR (KBr) υ/cm−1: 2946, 2866, 1687, 1597, 1485, 1452, 1242, 1061, 834, 754. GC-MS m/z: 270 [M]+. Anal. found: C 70.89, H 4.97%; calcd for C16H14O4: C 71.10, H 5.22%.
°C. 1H-NMR (300 MHz, [D6]DMSO)
δ: 3.207 (s, 4H, –SCH2CH2S–), 6.948–7.663 (m, 16H, aromatic H), 8.913 (s, 2H, –CHN), 12.984 (s, 2H, –OH). 13C-NMR (300 MHz, [D6]DMSO)
δ: 30.6 (–SCH2CH2S–), 163.0 (–CHN), 116.6–160.2 (aromatic C). FTIR (KBr) υ/cm−1: 3000, 1612, 1573, 1561, 1468, 1279, 1181, 907, 753. UV-Vis (CH2Cl2) λmax/nm (ε/mol−1 L cm−1): 351 (26900), 272 (52100), 234 (54800). FAB-MS m/z: 485 [M]+ (70%). Anal. found: C 69.82, H 4.91, N 5.38, S 12.74%; calcd for C28H24N2O2S2: C 69.39, H 4.99, N 5.78, S 13.23%.
°C. 1H-NMR (300 MHz, [D6]DMSO)
δ: 3.195 (s, 4H, –SCH2CH2S–), 7.023–8.522 (m, 20H, aromatic H), 9.630 (s, 2H, –CHN), 15.384 (s, 2H, –OH). 13C-NMR (300 MHz, [D6]DMSO)
δ: 30.0 (–SCH2CH2S–), 155.9 (–CHN), 118.4–136.3 (aromatic C). FTIR (KBr) υ/cm−1: 3400, 1620, 1553, 1460, 1326, 1168, 816, 743. UV-Vis (DMSO) λmax/nm (ε/mol−1 L cm−1): 471 (18900), 439 (25700), 398 (36800), 320 (35700). FAB-MS m/z: 585 [M]+ (100%). Anal. found: C 73.07, H 4.66, N 4.64%; calcd for C36H28N2O2S2: C 73.95, H 4.83, N 4.79%.
°C. 1H-NMR (300 MHz, CDCl3)
δ: 4.519 (s, 4H, –OCH2CH2O–), 6.737–8.145 (m, 16H, aromatic H), 9.083 (s, 2 H, –CHN). FTIR (KBr) υ/cm−1: 3423, 1619, 1593, 1489, 1449, 1374, 1221, 1047, 748. UV-Vis (DMSO) λmax/nm (ε/mol−1 L cm−1): 391 (16750), 354 (36800), 323 (30050). FAB-MS m/z: 453 [M]+ (65%). Anal. found: C 73.90, H 5.06, N 5.74%; calcd for C28H24N2O4: C 74.32, H 5.35, N 6.19%.
°C. 1H-NMR (300 MHz, [D6]DMSO)
δ: 5.572–5.889 (s, 3H, –SCHCH2), 6.423–8.476 (m, 16H, aromatic H), 8.805 (s, 1H, –CHN), 8.934 (s, 1H, –CHN). 13C-NMR (300 MHz, [D6]DMSO)
δ: 159.4 (–CHN), 162.2 (–CHN), 113.5–166.0 (aromatic C and –CHCH2). FTIR (KBr) υ/cm−1: 1604, 1573, 1519, 1525, 1435, 1249, 1177, 1143, 927, 747. UV-Vis (DMSO) λmax/nm (ε/mol−1 L cm−1): 475 (10800), 440 (10700), 285 (57800). FAB-MS m/z: 541 [M]+ (85%). Anal. found: C 62.32, H 3.75, N 5.00, S 11.31%; calcd for C28H21CoN2O2S2: C 62.22, H 3.92, N 5.18, S 11.86%.
°C. FTIR (KBr) υ/cm−1: 1604, 1570, 1518, 1438, 1388, 1139, 755. UV-Vis (DMSO) λmax/nm (ε/mol−1 L cm−1): 879 (26), 567 (142), 455 (9300), 309 (10040). FAB-MS m/z: 541 [M]+ (100%). Anal. found: C 61.73, H 3.89, N 4.95%; calcd for C28H22NiN2O2S2: C 62.13, H 4.10, N 5.18%.
>250°C. 1H-NMR (300 MHz, [D6]DMSO)
δ: 6.364–7.682 (m, 16H, aromatic H), 8.708 (s, 2H, –CHN). FTIR (KBr) υ/cm−1: 1611, 1560, 1465, 1370, 1276, 1180, 752. FAB-MS m/z: 548 [M]+ (15%). Anal. found: C 61.10, H 4.29, N 5.25%; calcd for C28H22N2O2S2Zn: C 61.37, H 4.05, N 5.11%.
°C. 1H-NMR (300 MHz, [D6]DMSO)
δ: 5.574–5.957 (s, 3H, –SCHCH2), 6.621–8.583 (m, 20H, aromatic H), 9.347 (s, 1H, –CHN), 9.384 (s, 1H, –CHN). 13C-NMR (300 MHz, [D6]DMSO)
δ: 126.9 (–SCHCH2), 128.8 (– SCHCH2), 153.0 (–CHN), 155.3 (–CHN), 115–137 (aromatic C). FTIR (KBr) υ/cm−1: 1600, 1572, 1532, 1515, 1423, 1385, 1183, 747. UV-Vis (DMSO) λmax/nm (ε/mol−1 L cm−1): 510 (15500), 480 (20100), 446 (17100), 341 (33700), 284 (88400). FAB-MS m/z: 641 [M]+ (100%). Anal. found: C 67.15, H 3.86, N 4.15%; calcd for C36H25CoN2O2S2: C 67.50, H 3.93, N 4.37%.
°C. FTIR (KBr) υ/cm−1: 1608, 1569, 1530, 1389, 1266, 1177, 828, 747. UV-Vis (DMSO) λmax/nm (ε/mol−1 L cm−1): 881 (66), 599 (276), 471 (56700), 445 (43800), 334 (46500), 279 (91400). FAB-MS m/z: 641 [M]+ (100%). Anal. found: C 66.93, H 4.37, N 3.97%; calcd for C36H26NiN2O2S2: C 67.41, H 4.09, N 4.37%.
°C. 1H-NMR (300 MHz, [D6]DMSO)
δ: 6.566–8.229 (m, 20H, aromatic H), 9.363 (s, 2H, –CHN). FTIR (KBr) υ/cm−1: 1608, 1567, 1531, 1394, 1362, 1177, 832, 747. FAB-MS m/z: 648 [M]+ (85%). Anal. found: C 66.90, H 3.90, N 4.15%; calcd for C36H26N2O2S2Zn: C 66.72, H 4.04, N 4.32%.
°C. FTIR (KBr) υ/cm−1: 1598, 1583, 1469, 1390, 1305, 1284, 1141, 734. UV-Vis (DMSO) λmax/nm (ε/mol−1 L cm−1): 446 (26700), 312 (33700). FAB-MS m/z: 509 [M]+ (10%). Anal. found: C 65.90, H 4.43, N 5.25%; calcd for C28H22CoN2O4: C 66.02, H 4.35, N 5.50%.
°C. FTIR (KBr) υ/cm−1: 1600, 1583, 1469, 1279, 1241, 742, 500. UV-Vis (DMSO) λmax/nm (ε/mol−1 L cm−1): 908 (50), 624 (230), 471 (33950), 343 (19600), 314 (36350). FAB-MS m/z: 509 [M]+ (45%). Anal. found: C 65.54, H 4.81, N 5.06%; calcd for C28H22NiN2O4: C 66.05, H 4.36, N 5.50%.
°C. 1H-NMR (300 MHz, [D6]DMSO)
δ: 4.384 (s, 4H, –OCH2CH2O–), 6.293–7.778 (m, 16H, aromatic H), 8.774 (s, 2H, –CHN). FTIR (KBr) υ/cm−1: 1598, 1486, 1451, 1290, 1256, 752. FAB-MS m/z: 516 [M]+ (40%). Anal. found: C 64.89, H 4.34, N 5.29%; calcd for C28H22N2O4Zn: C 65.19, H 4.30, N 5.43%.
Structure determination
Single crystal X-ray diffraction data were collected for all the compounds on a Nonius Kappa CCD diffractometer in the ϕ scan+ω scan mode using Mo-Kα of wavelength 0.71073 Å. All the structures were solved using direct methods with SIR-92 (H2L1 and 3) and SHELXS-97 (H2L3, 1, 5, 7, 8 and 9) and were refined using the SHELXL-97 program.7,8 The diagrams were generated using ORTEP3.9 Full-matrix least-squares refinement with anisotropic thermal parameters for all non-hydrogen atoms was used. In case of the ligand structures, –OH hydrogen positions were recovered from the difference Fourier map. The hydrogen atoms were treated as riding atoms with fixed thermal parameters. The empirical absorption correction was applied in the case of 1 and 3. In other structures there was no absorption correction employed. The crystals are found to be stable and other details of data collection and structure refinement are provided in Table 4.†
| Compounds | H2L1 | H2L3 | 1 | 3 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| Empirical formula | C28H24N2O2S2 | C14H12NO2 | C28H21CoN2O2S2 | C28H22N2O2S2Zn | C36.5H28N2O2.5S2Ni | C18H13NOS Zn0.5 | C16H15Co0.50NO4 | C16H15Ni0.5O4 | C15H15NO3Zn0.5 |
| Formula weight | 484.61 | 226.25 | 540.52 | 547.97 | 657.44 | 648.08 | 314.75 | 314.64 | 289.96 |
| T/K | 173(2) | 173(2) | 150(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Tetragonal | Triclinic | Monoclinic | Tetragonal | Tetragonal | Tetragonal |
| Space group | P21/c | Pbcn | Cc | P41 | P1 | C2/c | P42/n | P42/n | P42/n |
| a/Å | 10.716(1) | 17.254(1) | 15.279(1) | 11.383(1) | 10.750(1) | 22.994(1) | 13.929(2) | 13.925(2) | 13.925(2) |
| b/Å | 16.811(1) | 8.395(1) | 12.807(1) | 11.383(1) | 12.299(1) | 7.612(1) | 13.929(2) | 13.925(2) | 13.925(2) |
| c/Å | 13.097(1) | 15.994(1) | 12.191(1) | 18.347(1) | 12.656(1) | 17.980(2) | 14.231(1) | 14.181(3) | 14.181(3) |
| α/° | 84.38(1) | ||||||||
| β/° | 104.23(1) | 94.51(1) | 67.41(1) | 110.55(1) | |||||
| γ/° | 70.99(1) | ||||||||
| U/Å3) | 2287.0(3) | 2316.7(1) | 2378.1(3) | 2377.3(3) | 1460.0(2) | 2946.8(5) | 2761.1(1) | 2749.8(8) | 2749.8(8) |
| Z | 4 | 8 | 4 | 4 | 2 | 4 | 8 | 8 | 8 |
| μ/mm−1 | 0.263 | 0.087 | 0.927 | 1.239 | 0.848 | 1.012 | 0.680 | 0.765 | 0.939 |
| Reflections collected | 12704 |
9192 | 6743 | 8877 | 5901 | 4626 | 17104 |
18110 |
17541 |
| Independent reflections | 4014 | 1496 | 3542 | 3546 | 4324 | 2501 | 2620 | 2594 | 2496 |
| R int | 0.0281 | 0.0501 | 0.0317 | 0.0485 | 0.0357 | 0.0311 | 0.1015 | 0.1233 | 0.1048 |
|
R
1
[I>2σ(I)] |
0.0634 | 0.0660 | 0.0276 | 0.0375 | 0.0507 | 0.0339 | 0.0439 | 0.0746 | 0.0382 |
|
wR
2
[I>2σ(I)] |
0.1619 | 0.1532 | 0.0624 | 0.072 | 0.1099 | 0.0664 | 0.0924 | 0.1695 | 0.0708 |
| R 1 (all data) | 0.0744 | 0.0939 | 0.0303 | 0.0477 | 0.0750 | 0.0529 | 0.0787 | 0.1044 | 0.0756 |
| wR 2 (all data) | 0.1712 | 0.1711 | 0.0636 | 0.0737 | 0.1229 | 0.0726 | 0.1060 | 0.1863 | 0.0818 |
Acknowledgements
CPR acknowledges financial support from the Council of Scientific and Industrial Research (CSIR), New Delhi, and the Department of Science and Technology, New Delhi. GR acknowledges an SRF fellowship from CSIR.References
- (a) S. Zhou, S. Liu and Z. Zhou, Huaxue Shiji, 2001, 23, 26 Search PubMed; (b) S. Pal, S. Ghosh, G. Mukherjee and A. K. Nandi, Polyhedron, 1998, 17, 3439 CrossRef CAS; (c) P. Chakraborty, S. K. Chandra and A. Chakravorty, Inorg. Chim. Acta, 1995, 229, 477 CrossRef CAS; (d) P. Chattopadhyay and C. Sinha, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal Chem., 1995, 76, 34; (e) K. Dey, D. Bandyopadhyay, K. K. Nandi, S. N. Poddar, G. Mukhopadhyay and G. B. Kauffman, Synth. React. Inorg. Met.-Org. Chem., 1992, 22, 1111 CAS; (f) E. F. W. Thurston, J. Photogr. Sci., 1990, 38, 34 Search PubMed; (g) H. P. Nur, M. T. Rahman and A. Malek, Inorg. Nucl. Chem. Lett., 1981, 17, 133 Search PubMed; (h) N. E. MacKenzie, R. H. Thomson and C. W. Greenhalgh, J. Chem. Soc., Perkin Trans. 1, 1980, 12, 2923 RSC; (i) K. Motoda, H. Sakiyama, N. Matsumoto, H. Okawa and D. E. Fenton, J. Chem. Soc., Dalton Trans., 1995, 20, 3419 RSC; (j) P. Comba, A. Fath, T. W. Hambley, A. Kühner, D. T. Richens and A. Vielfort, Inorg. Chem., 1998, 37, 4389 CrossRef CAS; (k) W.-X. Zhang, C. Ma and S. Si, Transition Met. Chem., 2001, 26, 380 CrossRef CAS.
- G. Rajsekhar, C. P. Rao, P. K. Saarenketo, E. Kolehmainen and K. Rissanen, Inorg. Chem. Commun., 2002, 5, 649 CrossRef CAS.
- (a) R. D. Cannon, B. Chiswell and L. M. Venanzi, J. Chem. Soc. A, 1967, 8, 1277 RSC; (b) F. P. Dwyer, N. S. Gill, E. C. Gyarfas and F. Lions, J. Am. Chem. Soc., 1954, 76, 383 CrossRef CAS; (c) G. Mukherjee, S. P. Poddar and K. Dey, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal Chem., 1986, 275, 25; (d) G. Mukherjee, S. P. Poddar, K. Choudhury and K. Dey, Transition Met. Chem., 1988, 13, 58 CAS.
- (a) K. Umakoshi, T. Yamasaki, A. Fukuoka, H. Kawano, M. Ichikawa and M. Onishi, Inorg. Chem., 2002, 41, 4093 CrossRef CAS; (b) X. Fan, R. Cao, M. Hong, W. Su and D. Sun, J. Chem. Soc., Dalton Trans., 2001, 20, 2961 RSC; (c) T. Arliguie, C. Lescop, L. Ventelon, P. C. Leverd, P. Thuery, M. Nierlich and M. Ephritikhine, Organometallics, 2001, 20, 3698 CrossRef CAS; (d) G. E. M. Mullen, P. J. Blower, D. P. Price, A. K. Powell, M. J. Howard and M. J. Went, Inorg. Chem., 2000, 39, 4093 CrossRef CAS; (e) T. Morikita, M. Hirano, A. Sasaki and S. Komiya, Inorg. Chim. Acta., 1999, 291, 341 CrossRef CAS; (f) L. R. Hanton and K. Lee, Inorg. Chem., 1999, 38, 1634 CrossRef CAS; (g) M. Hong, W. Su, R. Cao, F. Jiang, H. Liu and J. Lu, Inorg. Chim. Acta, 1998, 274, 229 CrossRef CAS; (h) J. G. Planas, M. Hirano and S. Komiya, Chem. Lett., 1998, 2, 123 CrossRef; (i) P. Chakroborty, S. K. Chandra and A. Chakravorty, Inorg. Chem., 1994, 33, 4959 CrossRef and references cited therein; (j) S. Y. Darensbourg, Heterolytic and homolytic S–C bond cleavage reactions in nickel thioether complexes, Book of Abstracts, 211th ACS National Meeting, New Orleans, LA, 1996 Search PubMed; (k) P. Chakraborty, S. Karmakar, S. K. Chandra and A. Chakravorty, Inorg. Chem., 1994, 33, 816 CrossRef CAS; (l) P. Chakraborty, S. K. Chandra and A. Chakravorty, Organometallics, 1993, 12, 4726 CrossRef CAS.
- (a) A. B. Charette, J. F. Marcoux and F. Belanger-Gariepy, J. Am. Chem. Soc., 1996, 118, 6792 CrossRef CAS; (b) A. K. Sah, C. P. Rao, E. K. Wegelius, E. Kolehmainen and K. Rissanen, Carbohydr. Res., 2001, 336, 249 CrossRef CAS; (c) A. D. Watson, C. P. Rao, J. R. Dorfman and R. H. Holm, Inorg. Chem., 1985, 24, 2820 CrossRef CAS; (d) M. L. Helm, C. M. Combs, D. G. VanDerveer and G. J. Grant, Inorg. Chim. Acta, 2002, 338, 182 CrossRef CAS.
- (a) J. C. Duff and E. J. Bills, J. Chem. Soc., 1934, 1305 RSC; (b) R. W. Hay, P. M. Gidney and G. A. Lawrance, J. Chem. Soc., Dalton Trans., 1975, 779 RSC; (c) P. A. Duckworth, F. S. Stephens, K. P. Wainwright, K. D. V. Weerasuria and S. B. Wild, Inorg. Chem., 1989, 28, 4531 CrossRef CAS.
- SIR-92: A program for crystal structure solution. A. Altomore, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 Search PubMed.
- G. M. Sheldrick, SHELX-97 Programs for Crystal Structure Analysis (Release 97-2), University of Göttingen, Germany, 1998 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
Footnote |
| † CCDC reference numbers: 172099 (H2L1, EGEXIX), 216935 (H2L3), 172100 (1, EGIXIB), 195200 (3, EGIXOH), 216930 (5), 216931 (6), 216932 (7), 216933 (8) and 216934 (9). See http://www.rsc.org/suppdata/nj/b3/b305313j/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |