Neutral and zwitterionic group 4 metal alkyls with ancillary boroxide ligands
Sarah C.
Cole
,
Martyn P.
Coles
* and
Peter B.
Hitchcock
The Department of Chemistry, University of Sussex, Falmer, Brighton, UK BN1 9QJ. E-mail: m.p.coles@sussex.ac.uk; Fax: 01273 677196; Tel: 01273 877339
First published on 7th October 2004
Abstract
The boroxide anion, [OB(mes)2]−, has been applied as an ancillary ligand to generate electron-deficient group 4 metal alkyls; however, the enhanced electrophilicity results in formation of tight ion-pairs in solution.
The quest for alternatives to the ubiquitous cyclopentadienyl anion in early transition-metal olefin polymerisation catalysis remains one of the most intensely studied areas of organometallic chemistry.1 As such, numerous ligand sets have been investigated as ancillary groups for this reactivity and many elegant approaches have been developed which enable fine-tuning of their steric and electronic properties. The chelating diamido framework ranks amongst the most successful group of ligands in this context, where, in addition to the nature of the linking-group, the N-substituents are highly influential in controlling the environment about the active-site. To reduce electron-density at the metal by decreasing the extent of π-donation, commonly associated with an increased activity, Schrock and co-workers incorporated boryl-groups adjacent to the donor nitrogen atom.2 Unfortunately, the effect was too pronounced in these cases and resulted in the formation of low-activity systems for olefin polymerisation, due to facile decomposition pathways for the neutral dimethyl precursors and strong anion binding in the cationic systems.
Recent work from our group3 has demonstrated that alkoxide ligands substituted with boryl groups (referred to as boroxides),4,5 have similar π-donor properties to substituted aryloxide ligands, illustrated by solid- and solution-state studies of transition-metal imido species. Since conventional aryloxides have been moderately successful as ancillary ligands in active catalysts for the polymerisation of α-olefins,6,7 we have initiated a study of group 4 metal alkyls incorporating boroxide ligands and their applications in ethylene polymerisation catalysis.
Alkyl-elimination reactions, employing dimesitylborinic acid as the ligand source, were investigated as a straightforward method for the synthesis of target compounds with general formula M{OB(mes)2}2R2 [M = group 4 metal, R = Me, CH2Ph].8 Reaction time was found to be critical when employing M(CH2Ph)4 as the metal reagent, and allowing a reaction to stir for extended periods frequently resulted in the introduction of a greater number of boroxide ligands than warranted by the stoichiometry of the reaction. It was established that reacting a 2∶1 and 3∶1 ratio of (mes)2BOH and Ti(CH2Ph)4 for 3 h and 15 h, respectively, proved optimal for the synthesis of Ti{OB(mes)2}2(CH2Ph)2 (1) and Ti{OB(mes)2}3(CH2Ph) (2).† Identical conditions employing Hf(CH2Ph)4, however, resulted in isolation of the tris- and tetrakis-boroxides Hf{OB(mes)2}3(CH2Ph) (3) and Hf{OB(mes)2}4 (4) rather than the anticipated analogues.
The tris(boroxide) mono(benzyl) compound 2 crystallised as the monomeric species,‡ with three terminal boroxide ligands and the benzyl group forming a distorted tetrahedral array about the central titanium atom (Fig 1). The Ti–O distances [av. 1.794 Å] are notably shorter than in the related d0-titanium boroxide, Ti(NtBu){OB(mes)2}2(Py)2,3 [av. 1.930 Å], although the validity of direct comparison is tempered by the presence of donor pyridine ligands and the increased coordination number in the imido complex. However, despite the formal low electron count for Ti in compound 2, there is no evidence for any η2-type interaction with the benzyl group [Ti⋯C56 = 2.942 Å; Ti–C55–C56 = 109.98(18)°].
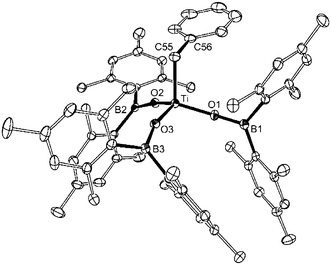 | ||
| Fig. 1 Molecular structure of 2 (ellipsoids drawn at the 20% probability level, hydrogen atoms omitted). Selected bond lengths (Å) and angles (°): Ti–O1 1.7965(18), Ti–O2 1.7923(18), Ti–O3 1.7918(19), Ti–C55 2.081(3); Ti–O1–B1 162.57(19), Ti–O2–B2 163.6(3), Ti–O3–B3 160.60(18), Ti–C55–C56, 109.98(18). | ||
A number of substituted phenoxide ligands have been shown to form monomeric compounds with formula Ti(OAr)4 [Ar = 2,6-iPr2C6H3,9 2-tBuC6H4 and 2,3,5,6-Me4C6H10] while the homoleptic siloxides M(OSiR3)4 [R = OtBu,11,12 Ph13] are known for all members of the group. The analogous tetra(boroxide), Hf{OB(mes)2}4 (4), also crystallises as the monomeric species‡ (Fig. 2), contrasting with the only other heteroleptic boroxide, [Fe{OB(mes)2}{μ-OB(mes)2}]2, which contains both terminal and bridging boroxides.5 The intra-ligand angles at hafnium [range 107.9(3)–111.7(4)°] deviate only slightly from ideal tetrahedral geometry, suggesting a relatively unstrained system. This is likely due to the boron atom displacing the mesityl substituents from the metal centre which effectively reduces the cone angle of the ligand, and the interlocking, parallel orientation of the aryl moieties from different ligands (Fig 2b). The Hf–O bond lengths [1.902(7) and 1.916(7) Å] tend towards the low end of the range observed in the tetrasiloxides [1.935(4)–1.949(4) Å]11 and the tris(alkoxide) species, Hf{OAr}3Cl [Ar = 2,6-tBuC6H3: 1.917(3)–1.938(3) Å],14 although in the absence of solution-state data, these effects may derive from purely steric factors rather than be electronic in origin.
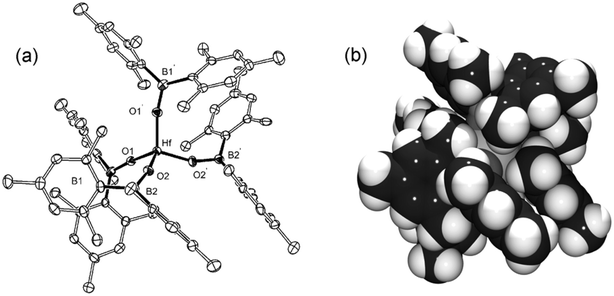 | ||
| Fig. 2 (a) Molecular structure of 4 (ellipsoids drawn at the 30% probability level, hydrogen atoms omitted). Selected bond lengths (Å) and angles (°): Hf–O1 1.902(7), Hf–O2 1.916(7); Hf–O1–B1 167.7(7), Hf–O2–B2 160.0(7). (b) Spacefill model of 4 (generated from the same projection). | ||
The bis(boroxide) species, Ti{OB(mes)2}2(CH2Ph)2 (1), constitutes the most promising precursor for the generation of [ML2R]+ cations, widely believed to be the active species in olefin polymerisation catalysis. The reaction between 1 and the well-defined borane activator, B(C6F5)3, generated a single new species (5), characterised spectroscopically in solution (Fig. 3). The 1H NMR spectrum showed the appearance of two signals corresponding to the CH2Ph protons [δ 3.54 and 3.39], each of which are shifted down-field from the corresponding singlet in the neutral dialkyl precursor [δ 2.85]. The higher frequency resonance appears as an unresolved multiplet which collapses to a broadened singlet in the 1H{11B} experiment, indicating that this resonance corresponds to a boron bound benzyl group (2JBH = ∼9 Hz). We attribute this signal to the formation of the anion [B(C6F5)3(CH2Ph)]−, commensurate with the desired alkyl abstraction process. The aromatic resonances associated with this component appear at higher field than observed for the free ion (Fig. 3), indicative of an η6-interaction between the titanium cation and the boroxide anion.15 Such interactions have previously been observed for group 4 systems employing aryloxide ligands,6,7 further demonstrating the similarity between the two ligand systems.
![1H NMR spectrum (C6D6, 300 MHz) of [Ti{OB(mes)2}2(CH2Ph)][B(C6F5)3(CH2Ph)]
(5) highlighting resonances corresponding to the η6-bound anion.](/image/article/2004/DT/b414155e/b414155e-f3.gif) | ||
| Fig. 3 1H NMR spectrum (C6D6, 300 MHz) of [Ti{OB(mes)2}2(CH2Ph)][B(C6F5)3(CH2Ph)] (5) highlighting resonances corresponding to the η6-bound anion. | ||
The ability of cationic metal systems to polymerise α-olefins is influenced by the strength of the cation–anion interaction, where displacement of the latter from the coordination sphere is generally necessary before propagation is able to proceed. Given the observed formation of the ion-pair in solution described above, it was perhaps unsurprising that exposure of a toluene solution of 5 to ethylene (7.5 bar) did not result in the formation of polyethylene. Additional complications with the aforementioned Ti/Zr alkoxide systems is the formation of a stable mono-insertion product with a range of unsaturated substrates, affording stable (inactive) cations.7 The possible formation of insertion products, in addition to the generation of cationic species from these well-defined precatalysts with alternative activators forms a part of our ongoing study into this area, and will be reported in due course.
We wish to thank Mr Gregory Knapp for the crystallisation of compound 2, and Dr A. G. Avent for helpful discussions with NMR data.
Notes and references
- G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428 CrossRef CAS; V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283 CrossRef CAS.
- T. H. Warren, R. R. Schrock and W. M. Davis, Organometallics, 1996, 15, 562 CrossRef CAS; T. H. Warren, R. R. Schrock and W. M. Davis, Organometallics, 1998, 17, 308 CrossRef CAS.
- S. C. Cole, M. P. Coles and P. B. Hitchcock, J. Chem. Soc., Dalton Trans., 2002, 4168 RSC.
- K. J. Weese, R. A. Bartlett, B. D. Murray, M. M. Olmstead and P. P. Power, Inorg. Chem., 1987, 26, 2409 CrossRef CAS.
- H. Chen, P. P. Power and S. C. Shoner, Inorg. Chem., 1991, 30, 2884 CrossRef CAS.
- M. G. Thorn, Z. C. Etheridge, P. E. Fanwick and I. P. Rothwell, Organometallics, 1998, 17, 3636 CrossRef CAS.
- M. G. Thorn, Z. C. Etheridge, P. E. Fanwick and I. P. Rothwell, J. Organomet. Chem., 1999, 591, 148 CrossRef CAS.
- S. C. Cole, M. P. Coles and P. B. Hitchcock, Organometallics, 2004 DOI:10.1021/om049520j; S. J. Birch, S. R. Boss, S. C. Cole, M. P. Coles, R. Haigh, P. B. Hitchcock and A. E. H. Wheatley, Dalton Trans., 2004 10.1039/b410945g.
- L. D. Durfee, S. L. Latesky, I. P. Rothwell, J. C. Huffman and K. Folting, Inorg. Chem., 1985, 24, 4569 CrossRef CAS; R. Minhas, R. Duchateau, S. Gambarotta and C. Bensimon, Inorg. Chem., 1992, 31, 4933 CrossRef CAS.
- R. T. Toth and D. W. Stephan, Can. J. Chem., 1991, 69, 172 CAS.
- K. W. Terry, C. G. Lugmair and T. D. Tilley, J. Am. Chem. Soc., 1997, 119, 9745 CrossRef CAS.
- M. P. Coles, C. G. Lugmair, K. W. Terry and T. D. Tilley, Chem. Mater., 2000, 12, 122 CrossRef CAS.
- B. F. G. Johnson, M. C. Klunduk, C. M. Martin, G. Sankar, S. J. Teate and J. M. Thomas, J. Organomet. Chem., 2000, 596, 221 CrossRef CAS.
- L. Chamberlain, J. C. Huffman, J. Keddington and I. P. Rothwell, J. Chem. Soc., Chem. Commun., 1982, 805 RSC; S. L. Latesky, J. Keddington, A. K. McMullen, I. P. Rothwell and J. C. Huffman, Inorg. Chem., 1985, 24, 995 CrossRef.
- C. Pellecchia, A. Grassi and A. Immirzi, J. Am. Chem. Soc., 1993, 115, 1160 CrossRef; M. Bochmann, S. J. Lancaster, M. B. Hursthouse and K. M. Abdul Malik, Organometallics, 1994, 13, 2235 CrossRef.
Footnotes |
| † Selected spectroscopic data (C6D6, 298 K): 1, 1H NMR: δ 6.95 (t, 4H, C6H5), 6.82 (t, 4H, C6H5), 6.80 (s, 8H, C6H2), 6.72 (s, 2H, C6H5), 2.85 (s, 4H, CH2), 2.28 (s, 24H, 2,6-Me2), 2.26 (s, 12H, 4-Me). 13C NMR: δ 142.7 (C), 141.1 (CH), 138.8 (CH), 129.0 (CH), 128.8 (C), 128.0 (C), 124.5 (CH), 93.2 (CH2), 23.0 (CH3), 21.4 (CH3). 2, 1H NMR: δ 6.87 (d, 2H, C6H5), 6.78 (t, 1H, C6H5), 6.65 (s, 12H, C6H2), 6.52 (d, 2H, C6H5), 3.18 (s, 2H, CH2), 2.25 (s, 36H, 2,6-Me2), 2.12 (s, 24H, 4-Me). 13C NMR: δ 142.7 (C), 141.0 (CH), 138.7 (CH), 129.0 (CH), 128.9 (C), 128.8 (C), 124.4 (CH), 93.2 (CH2), 23.0 (CH3), 21.4 (CH3). 4, 1H NMR: δ 6.62 (s, 16H, C6H2), 2.21 (s, 48H, 2,6-Me2) 2.12 (s, 24H, 4-Me). 13C NMR: 140.6 (C), 139.4 (br C), 138.2 (C), 127.3 (CH), 22.6 (CH3), 21.3 (CH3). 5, 1H NMR: δ 7.02 (t, 2H, TiCH2Ph), 6.98 (d, 2H, TiCH2Ph), 6.73 (t, 1H, TiCH2Ph), 6.65 (s, 8H, C6H2), 6.45 (d, 2H, BCH2Ph), 6.19 (t, 1H, BCH2Ph), 5.71 (t, 2H, BCH2Ph), 3.54 (br q, 2H CH2), 3.39 (s, 2H CH2), 2.15 (s, 24H 2,6-Me2), 2.08 (s, 12H, 4-Me). 13C NMR: δ 149.6 (C), 145.9 (C), 140.1 (C), 140.6 (d, C6F5), 138.5 (d, C6F5), 136.5 (d, C6F5), 136.2 (o-BCH2Ph), 129.2 (m-C6H2), 129.1 (p-BCH2Ph), 128.9 (m-TiCH2Ph), 128.3 (o-TiCH2Ph), 127.9 (p-TiCH2Ph), 124.0 (m-BCH2Ph), 98.3 (TiCH2Ph), *, 22.8 (C6H2Me3), 21.2 (C6H2Me3) [* resonance for BCH2Ph was not observed, presumably due to quadrupolar broadening]. |
‡ Crystallographic data for 2: C61H73B3O3Ti·(C5H12), M
= 1006.67, T
= 173(2) K, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No. 2), a
= 14.0992(7), b
= 14.5950(7), c
= 17.1898(9)
Å, α
= 93.847(2), β
= 95.609(2), γ
= 118.391(2)°, U
= 3070.8(3)
Å3, Z
= 2, Dc
= 1.09 Mg m−3, μ
(Mo–Kα)
= 0.18 mm−1, independent reflections = 10802 (Rint
= 0.053), R1 [for 7204 reflections with I > 2σ(I)]
= 0.062, wR2 (all data)
= 0.163. Crystallographic data for 4: C72H88B4HfO4, M
= 1239.15, T
= 173(2) K, monoclinic, space group C2/c
(No. 15), a
= 23.147(2), b
= 14.8478(7), c
= 21.429(2)
Å, β
= 119.350(3), U
= 6419.4(9)
Å3, Z
= 4, Dc
= 1.28 Mg m−3, μ
(Mo–Kα)
= 1.67 mm−1, independent reflections = 4414 (Rint
= 0.193), R1 [for 3036 reflections with I > 2σ(I)]
= 0.070, wR2 (all data)
= 0.141. CCDC reference numbers 250060 and 250061. See http://www.rsc.org/suppdata/dt/b4/b414155e/ for crystallographic data in CIF or other electronic format.
(No. 2), a
= 14.0992(7), b
= 14.5950(7), c
= 17.1898(9)
Å, α
= 93.847(2), β
= 95.609(2), γ
= 118.391(2)°, U
= 3070.8(3)
Å3, Z
= 2, Dc
= 1.09 Mg m−3, μ
(Mo–Kα)
= 0.18 mm−1, independent reflections = 10802 (Rint
= 0.053), R1 [for 7204 reflections with I > 2σ(I)]
= 0.062, wR2 (all data)
= 0.163. Crystallographic data for 4: C72H88B4HfO4, M
= 1239.15, T
= 173(2) K, monoclinic, space group C2/c
(No. 15), a
= 23.147(2), b
= 14.8478(7), c
= 21.429(2)
Å, β
= 119.350(3), U
= 6419.4(9)
Å3, Z
= 4, Dc
= 1.28 Mg m−3, μ
(Mo–Kα)
= 1.67 mm−1, independent reflections = 4414 (Rint
= 0.193), R1 [for 3036 reflections with I > 2σ(I)]
= 0.070, wR2 (all data)
= 0.141. CCDC reference numbers 250060 and 250061. See http://www.rsc.org/suppdata/dt/b4/b414155e/ for crystallographic data in CIF or other electronic format. |
| This journal is © The Royal Society of Chemistry 2004 |