Perylene bisimide dyes as versatile building blocks for functional supramolecular architectures
Frank
Würthner
*
Institut für Organische Chemie, Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: wuerthner@chemie.uni-wuerzburg.de; Fax: + 49-931-8884756
First published on 12th May 2004
Abstract
Perylene bisimide dyes and their organization into supramolecular architectures through hydrogen-bonding, metal ion coordination and π–π-stacking is discussed; further self-assembly leading to nano- and meso-scopic structures and liquid-crystalline compounds is also addressed.
Frank Würthner was born in 1964 in Villingen-Schwenningen (Germany). He studied chemistry at the University of Stuttgart where he received his Ph.D. degree in Organic Chemistry in 1993 working with Franz Effenberger. After a postdoctoral stay (1994/95) as an Alexander von Humboldt Fellow at MIT in Cambridge/MA (USA) in the laboratory of Julius Rebek, Jr. he spent two years at the BASF Central Research Laboratories in Ludwigshafen (Germany). In 1997 he joined the group of Peter Bäuerle at the University of Ulm where he received the venia legendi (Habilitation) in 2001. Since October 2002 he has been a full professor at the University of Würzburg. His research interests include merocyanine, perylene and porphyrin dye chemistry, noncovalent synthesis of nano- and meso-scopic structures, liquid crystals, and applications of organic materials in electronics and photonics. Recently he received the Arnold-Sommerfeld award of the Bavarian Academy of Science. |
Introduction
Perylene tetracarboxylic acid bisimide, in short perylene bisimide, based colorants have received considerable attention in academic as well as industrial dye and pigment research.1,2 Perylene bisimides were initially applied for industrial purposes as red vat dyes. After 1950 several members of the perylene bisimide family found high grade industrial applications as pigments (especially in automotive finishes) due to the favorable combination of insolubility and migrational stability, light- and weather-fastness, thermal stability and chemical inertness as well as high tinctorial strength with hues ranging from red to violet, and even black shades.2 More recent applications of perylene bisimide pigments are in the field of electronic materials, among which perylene bisimides are the best n-type semiconductors available to date.3,4 This n-type semiconductivity is related to the high electron affinity of rylene bisimide dyes,5 which makes naphthalene, perylene, as well as higher rylene bisimide dyes6 most promising for application in organic field effect transistors.7 In addition, based on their unique combination of optical, redox, and stability properties perylene bisimide dyes have already been investigated for more than a decade in electrophotography (xerographic photoreceptors)8 and photovoltaics.9However, owing to the intrinsic insolubility of perylene bisimides, only in 1959 was their potential as fluorescent dyes with high fluorescence quantum yield and photostability discovered10 which constituted a fertile soil for research on light-induced energy and electron transfer processes11–13 as well as more application-directed studies in the field of laser dyes and fluorescent light collectors.14–17 The past years have witnessed an ever increasing interest in this class of chromophores because of their above-mentioned favorable properties. Thus, perylene monoimide and bisimide dyes have proved to be the best fluorophores nowadays available for single molecule spectroscopy.18 Their ingenious incorporation into well-defined positions within shape-persistent dendrimers by Müllen and coworkers enabled unprecedented insight into dye–dye interactions on the single molecule level investigated by Hofkens, De Schryver and coworkers.19 In a further study, Adams and coworkers showed the suitability of perylene bisimide dyes for sensors with specificity at the single molecule level if appropriate receptor units are attached to the perylene bisimide fluorophore.20
Although the discussion of these covalent multichromophoric architectures and their properties would surely warrant review, this feature article focuses on another topic, namely, the supramolecular organization of perylene bisimide dyes to form complex functional architectures, a subject that was mainly studied in the author's laboratory. In this work the emphasis lies on the control of the spatial organization of perylene bisimide dyes by molecular-recognition-directed self-assembly. Such self-organization of functional dyes should be of great utility to tailor defined multichromophoric objects or bulk solid state materials with novel optical and electronic functionalities.
Available perylene bisimide dyes
Synthesis
Although insoluble and high-melting organic pigments like 1a–c could be easily obtained by the reaction of perylene tetracarboxylic acid bisanhydride with a multitude of aromatic and aliphatic amines, most topics of current interest require better soluble dyes. For the preparation of soluble perylene bisimide dyes two different strategies proved to be successful. The first one was developed by Langhals and coworkers, who ‘dissolved’ the pristine perylene bisimide chromophore by introducing solubilizing substituents at the imide nitrogen (1d, 1e).21 The dyestuffs obtained by this approach exhibit usually indistinguishable absorption and emission properties because nodes in the HOMO and LUMO at the imide nitrogen reduce the coupling between the perylene bisimide units and the imide substituents to a minimum.22 The second synthetically more elaborate strategy was to introduce substituents at the carbocyclic scaffold in the so-called bay-area. This approach was first mastered by Seybold and coworkers at BASF. They have incorporated four phenoxy groups in high yields by nucleophilic displacement of chlorine substituents (9a, Scheme 1).16 Introduction of other nucleophiles proved to be difficult and a single reaction product with reasonable yield was rarely afforded.23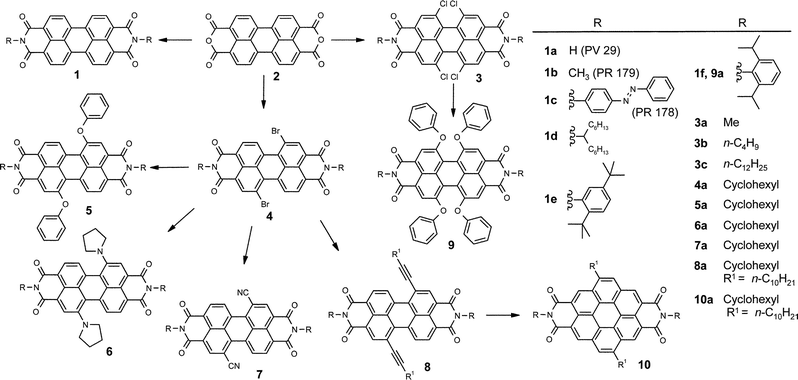 | ||
| Scheme 1 | ||
Although the procedure for fourfold chlorination of perylene bisanhydride 2 was already known for a while (even that it is not a trivial reaction due to contamination with three- and five-fold chlorination products),24,25 only very recently was it discovered that the bromination of perylene bisanhydride 2 affords disubstituted derivatives 4.26 However, the product mixture obtained by bromination is even worse than in the case of chlorination as threefold bromination products and significant amounts of a second dibromo regioisomer are formed. Moreover, the latter cannot be removed easily from the product mixture27 and is only detectable by high field (>400 MHz) 1H NMR spectroscopy.28 Gratifyingly, exchange of the bromine substituents of 4 is straightforward; thus, carbon,29 cyano,30 oxygen26 and nitrogen31 nucleophiles could be coupled to the perylene core leading to novel perylene bisimide dyes 5–8 with interesting optical and redox features. From the acetylenic derivative 8a even corenene bisimides 10 became available; however, also in this case an isomeric mixture, which could not be separated, was obtained.27
Optical properties
Since the discovery of the intense yellow-green photoluminescence of the parent perylene bisimides 1, numerous perylene bisimide dyes have been synthesized for their applicaton as fluorescent standards, in fluorescent light collectors, or as laser dyes.21 For such applications it has been proven to be very advantageous that the imide substituent has a negligible influence on the absorption and emission properties of perylene bisimides because of the nodes of the HOMO and LUMO orbitals at the imide nitrogens (Fig. 1). Therefore, perylene bisimides can be regarded as a closed chromophoric system with an S0–S1 transition (polarized along the extended molecular axis) whose intensity and position remain unaltered by the respective imide substituents.22 In addition, the coloristic properties of perylene bisimide dyes are scarcely dependent upon the environment, i.e. little solvatochromism is observed for these dyes. Accordingly, imide substituents are well-suited to tailor application-directed properties like solubility and to prevent aggregation that has a pronounced influence on the optical spectra. Very high solubility has been realized by Langhals's so-called swallow-tail-substituents as in derivative 1d. For the longer homologue with 1-nonyldecyl chains, solubility up to 35 g in 100 ml n-heptane has been reported.32 For most applications (such as coloration of plastics) twisted aryl groups like in 1e and 1f seem to be more advantageous since they not only increase the solubility but also prevent aggregation by sterical means.16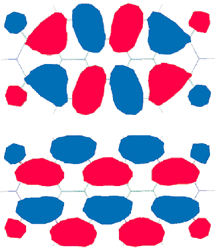 | ||
| Fig. 1 HOMO (top) and LUMO (bottom) of perylene bisimides. Note that both frontier orbitals exhibit nodes at the imide nitrogens. | ||
The data in Table 1 reveal that the absorption and emission maxima are almost identical for 1d and 1e (first two entries) and that the fluorescence quantum yield is quantitative. Because of the fact that the fluorescence quantum yield and lifetime of dye 1d was not affected by oxygen, this dye was suggested as a convenient standard for the determination of fluorescence quantum yields.33 If aryl substituents at the imide group are not fixed in an orthogonal conformation, however, the fluorescence quantum yield decreases. Thus, for a perylene bisimide bearing simple phenyl substituents at the imide nitrogens a quantum yield of 70% has been reported,25 and for more electron-rich alkoxyphenyl substituents the quantum yield dropped to <5%.34,35 The former effect has been attributed to vibronic motions (‘loose bolt effect’)34 and the latter one to a photoinduced electron transfer from the electron-rich aromatic to the electron-deficient perylene bisimide unit.35
| λ abs/nm | ε/M−1 cm−1 | λ em/nm | Φ f | τ f/ns | |
|---|---|---|---|---|---|
| a Note that values for compounds 4–8 and 10 have most likely been determined for mixtures of 1,6- and 1,7-substituted isomers. b Value determined for 3c, see ref. 37. c Values measured in toluene. | |||||
1d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 33 |
526 | 88000 |
533 | 1.00 | 4.0 |
|
1e14,15 |
526 | 95000 |
537 | 1.00 | 3.7 |
|
3b14,b |
524 | 42300b |
551 | 0.92 | |
|
4a27 |
526 | 57300 |
547 | ||
|
5a26 |
549 | 55000 |
57835 |
ca. 168 |
4.5 |
|
6a31,c |
686 | 46000 |
721 | 0.35 | 4.5 |
|
7a30 |
530 | 47000 |
545 | 1.00 | |
|
8a27 |
552 | 45000 |
571 | ||
|
9a17 |
573 | 45300 |
608 | >0.96 | 7.4 |
|
10a27 |
511 | 19700 |
517 | ||
More pronounced changes in the absorption and emission bands take place if the perylene bisimides are substituted at the aromatic core in the bay-area, i.e. the positions 1, 6, 7, and 12. With two phenoxy groups attached at positions 1 and 7 the absorption maximum shifts by about 20 nm and with four phenoxy groups by almost 50 nm compared to that of the unsubstituted dyes 1, and the color of the fluorescence changes to orange (5) and red (9). In both cases all favorable properties of these perylene fluorophores, namely, high fluorescence quantum yield, small solvent effects on the optical properties and high photostability are maintained. More pronounced spectral changes occur upon substitution at bay positions with two electron-donating pyrrolidino groups that affords the dyes 6 with a green color due to the bathochromic shift of 160 nm and emission in the infrared region. As this spectral shift is caused by charge transfer, pronounced solvatochromism is observed for these green dyes and their fluorescence quantum yield is decreased. By contrast, little spectral changes take place if electron-withdrawing substituents are attached at the bay positions (dyes 3, 4, 7). In the case of coronene bisimides 10, significant changes in the absorption spectra are observed that are not typical for perylene bisimide dyes. Accordingly, new electronic properties arise by expansion of the π-conjugated system orthogonal to the imide–imide axis.
Redox properties
The electrochemical properties of perylene bisimides have been investigated by several groups. Table 2 summarizes the data from Salbeck et al.,36 Wasielewski and coworkers30,31 and Würthner et al.35,37 that have been all recalculated to a common reference, i.e. the ferrocene/ferrocenium couple.| Solvent | E red (PBI−/PBI2−) | E red (PBI/PBI−) | E ox (PBI/PBI+) | E ox (PBI+/PBI2+) | |
|---|---|---|---|---|---|
| a Determined for compound 21b. b The oxidation potential for cp2Fe is taken as +0.52 V vs. SCE. c The first and second oxidation waves of 6a in dichloromethane are +0.03 and +0.35 V, respectively. d Determined for compound 21c. | |||||
|
1d36 |
CH3CN | −1.21 | −0.98 | +1.21 | Irrev. |
|
1e36 |
CH3CN | −1.15 | −0.93 | +1.25 | Irrev. |
|
3c37 |
CH2Cl2 | −1.07 | −0.87 | ||
|
535,a |
CH2Cl2 | −1.29 | −1.11 | +1.05 | |
|
6a31,b,c |
PrCN | −1.46 | −1.28 | +0.16 | +0.23 |
|
7a30 |
PrCN | −0.92 | −0.59 | ||
|
935,d |
CH2Cl2 | −1.25 | −1.09 | +0.88 | |
These data show that perylene bisimides are fairly electron-deficient dyes, which are easy to reduce and rather difficult to oxidize. For most compounds two reversible reduction and one reversible oxidation waves are found in cyclic voltammetry. Substituents in the bay-area have a pronounced effect on the respective redox potentials. Whereas the parent dyes 1 exhibit a first reduction potential that is already comparable to C60, substitution by chlorine and especially the cyano group leads to dyes that are strong oxidants. Thus, the redox potential of 7a is comparable to p-chloranil and radical anionic species can be observed already in the presence of amines, e.g. in technical grade DMF solution.30 With phenoxy electron-donor substituents reduction is disfavored by ca. 0.1 V, whereas a reversible oxidation wave appears at lower potential. For pyrrolidino compound 6a, even two reversible oxidation waves are observed and the reduction requires low potential. Accordingly, these dyes are the only ones in the series which cannot be considered as electron-poor.
The electron-deficient character of perylene bisimide dyes is also a prerequisite for the high photochemical stability of these dyes because photooxidation as the major destructive mechanism for dyestuffs is disfavored. On the other hand, the majority of these dyes are strong reductants in their photoexcited state. This property has been widely applied to establish long-lived charge-separated states in photoinduced electron transfer cascades by the research groups of Wasielewski11 and Lindsey.12 Chemoluminescence applications of perylene dyes have been demonstrated by Salbeck et al.36 as well as by Bard and coworkers.5
Structural properties
The parent perylene bisimides exhibit flat π-systems as confirmed by X-ray diffraction of several single crystals.38,39 According to the bond lengths observed in these crystals, perylene bisimides can be regarded as being composed of two naphthalene half units (marked in red in Fig. 2a), each of which is attached to an imide unit and connected to the other naphthalene unit by two C sp2–C sp2 single bonds. If the connecting bonds between the two half units are considered as single bonds it is not very surprising that sterical strain in the bay-area can lead to a propeller-like twisting of the two naphthalene half units. This is indeed the case as proven crystallographically for a tetrachloro-substituted perylene bisimide37 with a torsional angle of 37° and a tetraphenoxy-substituted diazadibenzoperylene derivative with a smaller angle of 25°.40 This distortion from planarity imposes considerable constraints in the packing of these dyes in the solid state as well as in molecular aggregates. The advantage of this distortion is that it affords much better soluble dyes. The solubility increase is very pronounced for the tetraphenoxy-substituted derivatives since the solubility of these derivatives benefits additionally by entropic reasons as the motional freedom of the four phenoxy arms (Fig. 2b) has to be frozen upon packing in the solid state. Thus, dye 9a exhibits a solubility exceeding 100 g L−1 in ethyl acetate at 20 °C.16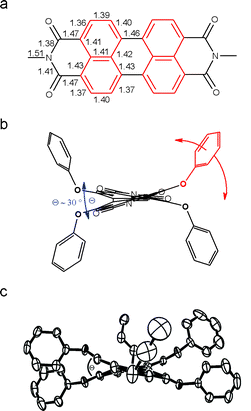 | ||
| Fig. 2 a) Bond lengths of 1b in the crystal;38 b) preferred conformation of the phenoxy substituents in bay-substituted perylene bisimides 9 in the gas phase according to AM1 calculations (and interpretation of the high solubility due to conformational mobility); and c) in the solid state according to a single crystal X-ray analysis. | ||
The conformational preference of tetraphenoxy-substituted perylene bisimides is still a matter of debate. From NMR studies it is evident that the barrier for planarization is small leading to rapid interconversion between the two twisted conformers and all attempts to isolate a particular enantiomer have failed so far.41 In the solid state a racemic mixture is observed and the phenoxy groups adopt an extended orientation that allows the most compact packing of these dyes (Fig. 2c).40 According to molecular dynamics and semiempirical geometry optimizations, however, the structure shown in Fig. 2b is preferred in the gas phase but several other conformational states exist within a small energy range.42 Accordingly, owing to solvent effects the preferred conformation or the conformational composition remains unclear in solution as well as in self-assembled architectures of these dyes.
Packing behavior in the crystal
The packing of perylene bisimides has been studied in great detail to understand and to optimize pigment colors for high grade applications including automotive coatings. Graser, Hädicke and Klebe of BASF have reported38 the crystal structures of 18 perylene bisimide pigments that contain the parent perylene bisimide skeleton 1 and that are only distinguished by their imide substituents. Six more derivatives have been reported recently by Zugenmaier et al.39 Crystal structures of the investigated dyes exhibit planar geometry of perylene bisimides, which are arranged in stacks showing a parallel orientation of the dyes at a distance between 3.34 and 3.55 Å (for comparison, a distance of 3.35 Å is found in graphite).38 For the stacking distance and the longitudinal and transverse offset between neighboring perylene bisimides (Fig. 3) steric requirements of the imide substituents seem to be of primary importance. As a result already for the series of simple alkyl chains significant changes in the two offsets have been found to enable interdigitation of alkyl chains at a larger distance of about 4.6 Å. For the ethyl and some benzyl substituents, rotational offsets have been observed leading to a screw-type stacking of the perylene bisimide dyes.38,39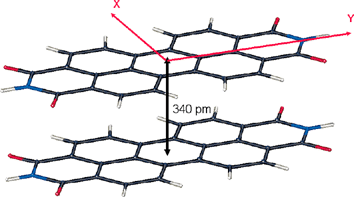 | ||
| Fig. 3 Most common π–π-stacking of perylene bisimides 1 in the solid state involving longitudinal and transverse offsets. | ||
One of the most remarkable features of these crystals is their crystallochromy, i.e. color changes resulting from the interaction of the π-systems in the crystal lattice. Thus, while all of these dyes exhibit an orange color and almost identical absorption spectra in solution, very different colors are found in the solid state depending on the packing of these dyes. Consequently, commercial pigments for red, maroon, red-violet, and even black are available based on perylene bisimides.2 The comprehensive study of Graser, Hädicke and Klebe38 enabled them to derive a rather simple empirical relationship for the dependence of the crystal color upon the longitudinal and transverse offsets of the dyes that relates the bathochromic shift and the band broadening to the extent of the π–π-contact area between the stacked chromophores (Fig. 4). Thus, already for quite small transverse offsets the π–π-interactions are brought to a minimum leading to pigments with little crystallochromic shifts compared with their solution spectra. Such optical properties have been observed for the dye 1c (Pigment Red 178) which exhibits a brilliant red shade. For smaller transverse shifts more perturbation takes place and maroon-type pigments such as 1b (Pigment Red 179) result. Owing to the more extended chromophoric system in the longitudinal direction, slipping in this direction is less effective in reducing the coupling between neighboring π-systems. As a consequence, pigments without or with only little transverse offset are often black due to the strongest electronic interaction between neighboring dyes (for example, Pigment Black 32). A more rigorous treatment of these crystallochromy effects has been given by Kazmaier and Hoffmann based on extended Hückel calculations on one-dimensional infinite stacks of dyes as a function of the two offset parameters.43 It is noteworthy that the last class of black pigments is considered to be of special interest for novel high technology applications where high exciton and/or charge carrier mobilities are desired, e.g. photoconductors, field effect transistors, or photovoltaic devices.
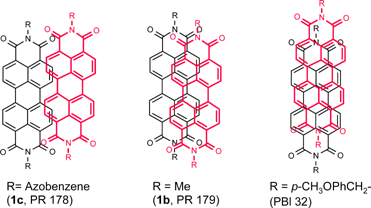 | ||
| Fig. 4 Transverse and longitudinal displacements of the stacked π-systems in the crystals of red (PR 178), maroon (PR 179) and black (PBI 32) perylene bisimide pigments. | ||
Self-assembly of perylene bisimide dyes
From the structural point of view, supramolecular assemblies constitute a state of matter that is just in between pigment particles (e.g.1a–c) and dissolved molecular dyes (e.g.1d–f). Accordingly, a reasonable approach to obtain supramolecular perylene bisimide dye assemblies is the modification of these chromophores in such a way that structural growth (or ‘self-assembly’) becomes possible only in one or two dimensions, while intermolecular interaction in a third dimension leading to crystalline solids is prevented by attaching appropriate substituents in the molecules. The judicious choice for such an approach is Seybold's dye 9a, in which the bulky phenoxy groups strongly interfere with aggregation of the π-systems as already expressed by remarkably high solubility. In this dye the imide substituents are properly positioned for the introduction of supramolecular receptor units. As a matter of fact, the most promising receptor units for supramolecular chemistry are particularly those which enable metal–ligand coordination and hydrogen-bonding as ubiquitously used in pigment chemistry! Thus, from the pigment chemist's point of view the chromophores are just modified in a way to prevent growth in all three spatial directions. On the other hand, a supramolecular chemist will discuss this approach in terms of applying receptor units to direct the self-assembly of the individual building blocks in a defined manner.Binding properties of receptor-functionalized perylene bisimides
Perylene bisimide dyes bearing one supramolecular receptor unit at one of the two imide functionalities could be synthesized and their complexation to complementary guests could be studied by titration experiments.44,45 For these studies it proved to be especially advantageous that for perylene bisimide building blocks different spectroscopic techniques could be applied that exhibit their highest sensitivity at different concentrations. Accordingly, not only binding constants in the lower range of 10–1000 M−1 could be determined by 1H NMR, but also higher binding constants up to 107 M−1 were accessible by UV/Vis and fluorescence spectroscopy due to spectral shifts or changes in the fluorescence quantum yield upon guest complexation. Scheme 2 shows examples for complex formation by properly functionalized perylene bisimide dyes through hydrogen-bonding and metal–ligand coordination, and Table 3 summarizes the binding constants determined in different solvents.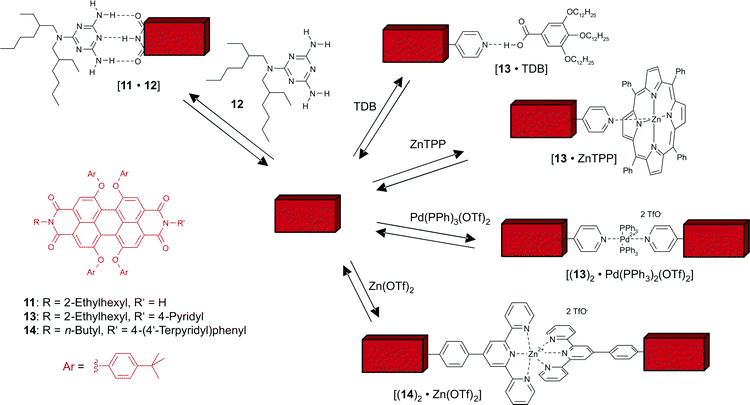 | ||
| Scheme 2 | ||
44,45
| Complex | Solvent | K/M−1 | −ΔG0298/kJ mol−1 | Method |
|---|---|---|---|---|
| a The reported values are microscopic site binding constants. b Isothermal titration calorimetry. | ||||
| [11·12] | [D8]THF | 15 | 6.5 | NMR |
| [11·12] | CDCl3 | 240 | 13.6 | NMR |
| [11·12] | CD2Cl2 | 270 | 14.0 | NMR |
| [11·12] | C6D6 | 1530 | 18.2 | NMR |
| [11·12] | CCl4 | 4080 | 20.6 | UV/Vis |
| [11·12] | MCH | 54000 |
27.0 | UV/Vis |
| [11·12] | n-Hexane | 90000 |
28.3 | Fluorescence |
| [13·TDB] | CDCl3 | 140 | 12.2 | NMR |
| [13·ZnTPP] | CDCl3 | 1650 | 18.3 | NMR |
| [(13)2·Pd(PPh3)2(OTf)2] | CDCl3 | 106a |
34a |
NMR |
| [(14)2·Zn(OTf)2] | CH3Cl/MeOH | >107a |
>40a |
ITCb |
The binding data in Table 3 for these complexation events in chloroform are in good accordance to other data known for the given receptor pairs. This result is not unexpected since the perylene bisimide unit is rather decoupled from the receptor units due to the previously mentioned nodes in the HOMO and LUMO orbitals at the imide nitrogens (the only exception is the imide receptor 11 with the hydrogen-bond accepting carbonyl groups). As an important consequence, the perylene bisimide emission remains unquenched for most coordination events including hydrogen-bonding and the given transition metal ions. Only in the case of ligation to ZnTPP is the perylene bisimide emission quenched due to the lower lying porphyrin S1 electronic state.
Owing to the high solubility of dye 11 the solvent effect on the strength of triple hydrogen-bonding in most organic solvents could be studied in detail.45 While this binding strength is very weak in hydrogen-bond acceptor solvents such as tetrahydrofuran and only moderate in chlorinated solvents, a dramatic increase in the binding constant was observed in the least polar aliphatic solvents. This leads to binding constants greater than 104 M−1 that are required once high degrees of association are desired at stoichiometric concentrations in the millimolar regime. Note that at a concentration of 1/K the degree of complexation amounts to 50% for a 1∶1 binding process at stoichiometric concentration of the two binding partners. For the self-assembly processes based on ditopic binding partners at stoichiometric concentrations K > 104 M−1 or preferably > 106 M−1 is mandatory and, therefore, the triple hydrogen-bonding motif will be only applicable in aliphatic solvents to construct large objects or nano- and meso-scopic assemblies. On the other hand, coordination of the pyridine receptor to Pt(II), Pd(II) or of the terpyridine receptor to Zn(II) ions is well-applicable for self-assembly in more polar media.
Although the ‘solvent polarity’ cannot be defined by a single parameter, a comparison of the characteristic binding constants depending on the solvent polarity is very illustrative. Thus, in Fig. 5 the solvent-dependent binding strengths for different noncovalent interactions are compared which are generally decreased with increasing solvent polarity. As K values higher than 106 are required to afford extended supramolecular assemblies in dilute solution (c < 1 mM), self-assembly through triple hydrogen-bonding can be achieved only in least polar, aliphatic solvents. However, under these conditions the π–π-stacking interactions also take place. Thus, for the π–π-aggregation of the parent perylene bisimides (left bar), higher interaction energies than for triple hydrogen-bonding are observed in polar as well as in nonpolar solvents (n-hexane, dichloromethane), whereas upon introduction of bay substituents the π–π-interaction is reduced. The synergistic effect of hydrogen-bonding and π–π-stacking may lead to interesting possibilities (vide infra). On the other hand, metal–ligand coordinative interactions are tunable to high binding strength even in polar solvents. Thus, the coordination of pyridine ligand to Pd(II) and Pt(II) ions is suitable to drive self-assembly processes to completion in CH2Cl2 or CHCl3 (in which the π-systems do not aggregate) in the millimolar regime. Clearly, from Fig. 5 experimental conditions can be chosen under which a particular recognition event takes place whereas another one is not yet active. In a follow-up process such as an increase of concentration or addition of a less polar solvent, however, the less favored interaction can also be triggered to enable structural growth in a hierarchical fashion.45
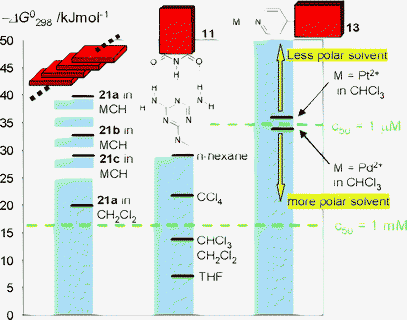 | ||
| Fig. 5 Comparison of the solvent dependence of noncovalent interactions, which have been applied for the self-organization of perylene bisimide dyes. On the left scale, Gibbs free energies for bimolecular association are given that have been calculated from the respective binding constants. Gibbs interaction energies of 34 kJ mol−1 are necessary to trigger self-assembly at micromolar concentration (50% of the receptors are complexed if both supramolecular building blocks are present at the same concentration), while only 17 kJ mol−1 are sufficient for self-assembly at millimolar concentration. | ||
If hydrogen-bonding receptor units are desired in more polar solvents, quadruple hydrogen-bonds may be applied to afford binding constants of about 108 M−1 in toluene.46 In a recent study, this motif was applied by Janssen and coworkers to form a supramolecular complex between a perylene bisimide (15) and an oligophenylenevinylene dye (16) (Scheme 3).47
 | ||
| Scheme 3 | ||
Metal–ligand-coordination-directed self-assembly in solution
The high binding strength of metal–ligand coordinative bonds in solvents where perylene bisimide dyes exhibit the highest solubility and the least tendency towards aggregation (see Fig. 5) makes this noncovalent interaction especially suited for self-assembly of defined multichromophoric nano-objects. Thus, several molecular squares could be prepared which incorporate perylene bisimide dyes bearing additional functional units such as ferrocene electrophores and pyrene chromophores (Scheme 4).48–50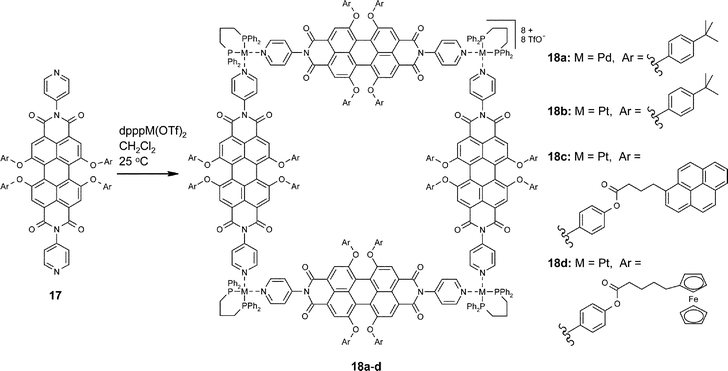 | ||
| Scheme 4 | ||
The dimension of these assemblies is about 2.4 × 2.4 nm with a large inner void that seems to be promising to accommodate additional guest molecules. Despite the large size and high molecular mass (almost 12 kD for 18c), electrospray ionization of intact squares was accomplished for 18b,c allowing unambiguous characterization of the assemblies in solution by Fourier transformation ion cyclotron resonance electrospray ionization (FTICR-ESI) mass spectrometry. Additional support for the exclusive existence of square-type oligomers comes from 1H and 31P NMR studies that show only a single product of high symmetry. Already the parent molecular square 18a exhibits remarkable functionality. Thus, the fluorescence quantum yield of the perylene bisimide fluorophore in the square remains almost unchanged as well as the electrochemical properties (two reversible reductions and one reversible oxidation for each perylene bisimide unit). For the ferrocene-substituted assemblies 18d all ferrocene units could be reversibly oxidized and a peak splitting in the reductive wave (‘spike effect’) was revealed by the cyclic voltammogramm.50 From the structural point of view, the pyrene-substituted assembly 18c seems to be a good model for the light-harvesting system of photosynthetic purple bacteria. The latter contain 24 chlorophyll and 8 carotene dyes arranged in a cyclic manner around an eightfold symmetry axis, whereas square 18c contains 16 pyrene and 4 perylene dyes organized around a fourfold symmetry axis. However, the photophysical functionality of the model system is quite distinct from the natural counterpart due to the competitive energy and electron transfer processes between the pyrene and perylene bisimide manifold.49
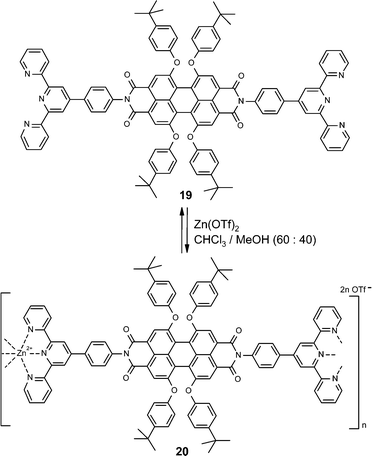
By employing the concept of metal-coordination-directed self-assembly, metallo-supramolecular polymers could be obtained through adding metal salts into a solution of the bis-terpyridine-functionalized perylene bisimide dye 19 according to Scheme 5. Although the coordinative bonds formed in this process are quite strong (compare Table 3) the appropriate choices of metal ion, i.e. zinc(II), and the solvent, i.e. chloroform/methanol, enables full reversibility of the binding event within a short timescale (<1 min). As in the case of metallo-supramolecular squares, also here the intense red photoluminescence of the incorporated perylene bisimide dye 19 was maintained in the polymeric product 20. The latter could be further deposited on a quartz substrate in a layer-by-layer approach by using the method of Decher.51
 | ||
| Scheme 5 | ||
π–π-Aggregation in solution
Although the stacking of the planar π-systems of perylene bisimide dyes is well documented in the crystal structures of the perylene pigments,38,39 only recently has the aggregation of these dyes been studied in solution and the Gibbs aggregation energies could be determined.35 For these studies it was useful to equip the imide functional groups with solubilizing 3,4,5-tridodecyloxyphenyl substituents to afford dissolution of these dyes in the least polar aliphatic solvents, in which the aggregation constants are strongest.Fig. 6a shows concentration-dependent UV/Vis studies for dyes 21a and 21c (see also Chart 1). Upon aggregation, both dyes experience significant spectral changes with defined isosbestic points. Evaluation of the absorbance changes depending upon the concentration in an isodesmic model (formation of one-dimensional aggregates with equal binding constants for the top and bottom faces of the dyes) by nonlinear regression analysis affords the aggregation constants and Gibbs association energies. The data for perylene bisimide dyes 21a–d bearing different substituents in the bay-area are compared in Table 4.
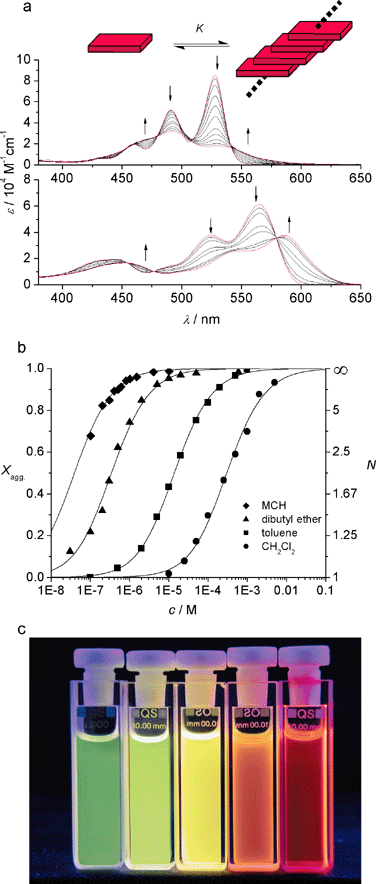 | ||
| Fig. 6 (a) Concentration-dependent UV/Vis spectra of 21a (top) and 21c (bottom): arrows indicate changes upon increasing concentration. (b) Fraction of aggregated π-faces Xagg and average number of molecules per aggregate N as a function of the concentration of 21a in different solvents. (c) Concentration-dependent luminescence of 21e in toluene. The concentrations from left to right are: 10−6, 10−5, 10−4, 10−3 , and 10−2 M. | ||
 | ||
| Chart 1 | ||
| K/M−1 | −ΔG0298/kJ mol−1 | |
|---|---|---|
| 21a | 1.5 × 107 | 40.9 |
| 21b | 6 × 105 | 33.1 |
| 21c | 1 × 105 | 29.0 |
| 21d | 700 | 16.2 |
The data in Table 4 clearly confirm that the π–π-stacking energy is significantly reduced by bay substituents. Thus, the highest Gibbs binding energy is obtained for the unsubstituted derivative 21a and the lowest value for the tetrachlorinated derivative 21d, the latter exhibits the most twisted π-system in this series.37 As the π–π-stacking energy is strongly reduced in more polar aromatic, chlorinated, and dipolar solvents, it is not surprising that for dyes 21b–d no evidence was obtained for aggregation up to the mM regime in these solvents. On the other hand, for the most strongly aggregating dye 21a, a recent study on solvent effects provided aggregation constants of 1.6 × 106 M−1 in dibutyl ether (εr = 3.0), 4.5 × 104 M−1 in toluene, and 1.6 × 103 M−1 in dichloromethane (εr = 8.9) (Fig. 6b). A comparison of these values with those reported in Table 3 shows that the intermolecular forces between unsubstituted perylene bisimide dyes are comparable with those of triple hydrogen-bonds (see also Fig. 5).
It is remarkable that dyes 21b,c show a bathochromic shift upon aggregation (Fig. 6a,b) leading to J-type aggregates. In general, for such aggregates the fluorescence intensity is not reduced in the aggregated state. This is also the case for 21b,c; however, these dyes suffer from reduced fluorescence quantum yields because of the electron-rich imide substituents, which may function as electron donors in a photoinduced electron transfer process (see above). This bathochromic shift of the absorption band can be explained in terms of a longitudinal displacement of the dyes toward each other in the aggregated state, an effect that has been attributed to a favorable stacking interaction between the electron-poor perylene bisimide core and the electron-rich trialkoxyphenyl units (compare the longitudinal offset in pigment black 32, Fig. 4). This interaction is also expressed by the magnitude of the binding constants since significantly higher values were observed for dyes 21 compared to those of related perylene bisimides with simple alkyl substituents at the imide nitrogens.
In contrast to the sterically restricted dyes 21b–d several possibilities arise for the π–π-stacking of dyes 21a and 21e with a fully planar π-system. For pigments (compare Fig. 3 and 4) longitudinal, transverse as well as rotational offsets have been observed.38,39 Apparently, all these displacements lead to attractive interactions between the π-systems. Therefore, in the pigment particles, it is the packing of the substituents at the imide nitrogen that controls the type and the degree of slipping of the cofacially aggregated dyes. For one-dimensional columnar aggregates in solution these packing restrictions are absent and, therefore, the aggregation of the perylene dyes should be only governed by the chromophore–chromophore interactions themselves. It is indeed quite interesting to see that spectral changes upon π–π-stacking of dyes 21a, 21e, and 22a as well as covalently tethered dyes 22b and 23 (Chart 2) are all quite similar irrespective of the substituents at the imide nitrogen and the solvent. Thus, in all cases where the flat parent perylene bisimides with only hydrogen substituents at the core are involved, a strong hypsochromy is observed pointing at H-type aggregation (Fig. 6a). However, it is not yet clear for this class of perylene bisimide dye aggregates what type of offset (longitudinal, transverse, or rotational) is given and whether indeed a common packing pattern is existing irrespective of the imide substituents. (Note that the illustrations presented in Fig. 6, and later in Scheme 6 and Scheme 7 depict only one reasonable possibility of how the dyes might be aggregated.) From the functional point of view, it is remarkable that the removal of electron-donating oxygens from the imide substituents in 21a leads to the strongly luminescent dye 21e (Φf = 0.7 in dichloromethane) and this luminescence changes from green (nonaggregated monomer) to yellow, orange, and even red upon aggregation (see Fig. 6c).
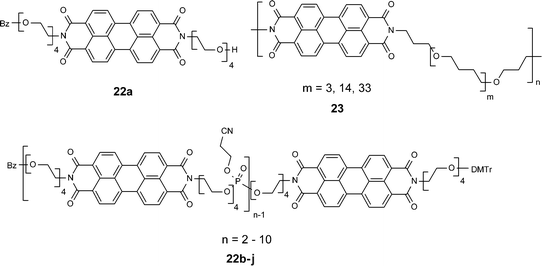 | ||
| Chart 2 | ||
Like in aliphatic solvents, π–π–stacking interactions between perylene bisimide dyes become strong in alcohols and water which are equally little polarizable solvents and, therefore, cannot solvate the π-surfaces well (this can easily be seen if the refractive indices of water, aliphatic, and alcoholic solvents are compared, which are only ca. 1.3). This fact has been applied by Li and coworkers52 and by Janssen's group to design foldable oligomers 22 and polymers 23 in which perylene bisimide units are connected by oligo(ethylene glycol) or poly(THF) chains that render the compounds soluble in an aqueous environment.53 For the monomeric model, compound 22a self-aggregation to one-dimensional stacks has been observed to be similar to that of compounds 21a–e and an aggregation constant of 52 M−1 in chloroform has been reported.54 Temperature-dependent studies confirmed that the aggregation process is an enthalpically driven process (ΔH° = −7.4 kcal mol−1; ΔS° = −17 cal mol−1 K−1). As in the case of 21e, also here quite remarkable changes in the optical properties arise leading to tunable luminescence colors.
Li's synthesis of higher oligomers is based on phosphoramidite chemistry and has been carried out in a sequential fashion to obtain defined molecules with up to 11 perylene bisimide units.52 In water, all of these molecules exist in a folded state that is characterized by a hypsochromically-shifted absorption band (H-aggregate). Also interesting here, emission colors from orange (dimers) to red (higher oligomers) arise which is quite surprising for dyes folded in the suggested H-type fashion. For electrostatic reasons, however, it can be assumed that the dyes are not really stacked perfectly cofacial on top of each other and, therefore, the forbidden lower energy excitonic transition may become allowed by means of a rotational displacement (compare also the stacking observed in the pigments). Upon increasing the concentration to >1 mM, further aggregation takes place that has been attributed to the formation of larger aggregates by self-assembly. Similar observations have been made by Janssen and coworkers on the related series of perylene bisimide–poly(THF) copolymers 23.53 In o-dichlorobenzene, these chromophores self-aggregate by folding to give H-type aggregates with almost identical spectral shifts in the UV/Vis and fluorescence spectra like those observed for 22. The fluorescence has been well characterized by time-resolved photoluminescence studies, and quantum yield determinations confirm that folding into the H-aggregated state does not fully quench but at least significantly reduces the fluorescence intensity.
Such folding behavior currently receives great interest as it is recognized that the formation of highly ordered structural domains distinguish natural macromolecules, i.e. proteins and DNA, from simple man-made technical polymers. In this context Li's recently reported hybrid structures consisting of alternating hydrophobic perylene bisimides and hydrophilic single-stranded desoxyribonucleic acids (ssDNA) connected by tetraethylene glycols seem to be particularly interesting. Depending on the sequence of the ssDNA linkers, unfolded or folded structures are accessible whose interaction with other DNA sequences may be monitored by fluorescence color changes caused by folding/unfolding of the perylene bisimide manifold.55 It is noteworthy that, indeed, folding of perylene bisimides takes place upon heating (an inverse temperature behavior) as shown in Scheme 6.
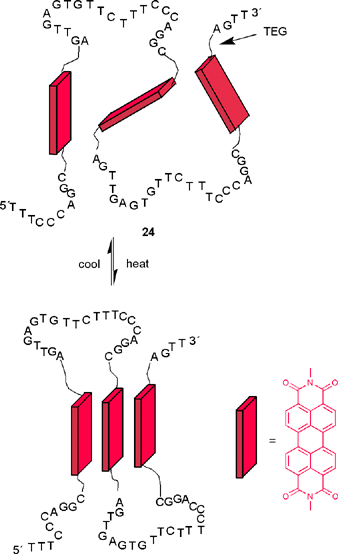 | ||
| Scheme 6 | ||
Perylene bisimide aggregates in the liquid-crystalline state
In the solid state perylene bisimide dyes 21a–e form liquid-crystalline (LC) phases over a broad temperature range. It is assumed that the π-systems pack in a similar way as observed for the dye aggregates in solution despite additional packing constraints that arise in the bulk of the LC phase. Credence to this assumption is given by wide angle X-ray diffraction data which show an increasing distance between the π-systems with increasing sterical demand of the substituents at the bay positions. Additionally, small angle X-ray diffraction data revealed a hexagonal columnar packing.35Considering the long history of academic and industrial research on perylene bisimide dye it is rather surprising that only in the last years have LC perylene bisimide dyes been discovered. The first observation of liquid-crystalline perylene bisimide dyes [1k,l and other derivatives with oligo(oxyethylene) chains (Chart 3)] was reported by Cormier and Gregg,56 subsequent to the discovery of liquid-crystallinity of the related perylene dyes 25 by Müllen, Spieß and coworkers.57 Poly(oxyethylene) substitutents at the imide nitrogens render the perylene bisimides 1k,l low-melting materials (e.g.1k melts at ca. 55 °C) that easily enter into a liquid-crystalline phase upon cooling from the melt. However, these LC phases are only monotropic and transformation into crystalline phases takes place upon standing.56 More recently, liquid-crystallinity of some coronene bisimides 10,27 perylene tetracarboxylic acid tetraesters 26,58,59 perylene bisimide 27,60 and even perylene bisimides 1g,h,i3 with simple aliphatic chains has been recognized. The last finding seems to be particularly surprising as the compounds have been known for a long time.
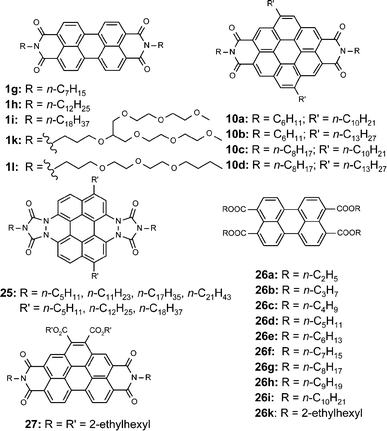 | ||
| Chart 3 | ||
Most of these dyes form hexagonal columnar mesophases as expected from their molecular structure. For 21a–c, Colho (ordered) and Colhd (disordered) phases are found which are stable from room temperature up to the clearing points between 283 and 373 °C.35 Because such a broad range for a mesophase is rather unusual it is assumed that solidification leads to a glassy solid which preserves the order of the liquid-crystalline phase. Likewise, 10a–d and 27 form Colho phases.27,60 However, as expected from their extended aromatic surfaces these compounds enter the mesophases only at considerably high temperatures (>180 °C) and recrystallize upon cooling. This property is prohibitive for applications at room temperature. Lower temperature columnar mesophases are accessible with tetracarboxylic acid esters 26 which can enter the LC phase at temperatures between 62 (26g) and 244 °C (26a) depending upon the alkyl substituents.59
For the parent compounds 1g–l the phase behavior is more complicated. For 1g–i it has been shown that several crystalline and liquid-crystalline phases occur between room temperature and the clearing points of these compounds.3 The liquid-crystalline phases exhibit high structural ordering in all three dimensions pointing at smectic layers with interdigitating alkyl chains in one dimension and columnar ordering with cofacially π–π-stacked perylene bisimide dyes in a second dimension. For 1k,l no X-ray data have been reported and, accordingly, no phase assignment could be made.56
Several of these compounds are promising for electronic applications since high n-type mobilities were measured in their liquid-crystalline phase (e.g. 0.11 cm2 V−1 s−1 for 1i and 0.08 cm2 V−1 s−1 for 21d),3,61 and electroluminescent diodes which emit yellow and orange light could be prepared (e.g. for 26 and 27).58,60 For organic light emitting diodes (OLEDs) such liquid-crystalline emitter materials could provide two distinct advantages. One is the preparation of an ordered aggregate by self-assembly from a molten liquid upon cooling between the electrodes, a process that seems to be far more elegant (and cheaper!) than traditional formation of crystalline films by vacuum sublimation. The second advantage is the higher brightness through emission of polarized light from the liquid-crystalline phase. However, these novel materials are not yet mature enough to match the efficiency of more developed crystalline or polymeric OLED materials.
Another challenging application of liquid-crystalline perylene bisimides is in the field of photovoltaics where these dyes may also exhibit a dual function, namely, light absorption and transport of charge carriers to the electrodes. Continuously over the last years Gregg and coworkers have explored the applicability of perylene bisimide dyestuffs as functional units for solar energy conversion. In Graetzel-type, dye-sensitized solar cells perylene bisimide dyes have been applied as efficient sensitizers of the inorganic semiconductor SnO2.62 In a more basic study thin polycrystalline films of the black perylene bis(phenethylimide) (the black color indicates strongly coupled dyes in the solid state, see above) have been shown to exhibit singlet exciton transfer lenghts of 2.5 μm which is the highest value ever reported for an organic material.63 In another study, n-type doping of 1l was achieved by adding 1 mol% of the radical anionic derivative of 28 leading to a highly conductive liquid-crystalline film (Scheme 7).64 By combining such favorable exciton and electron transport properties of perylene bisimide aggregates with a suitable p-type organic semiconductor in a bulk p–n-heterojunction material an efficient solar cell may be afforded (see below).
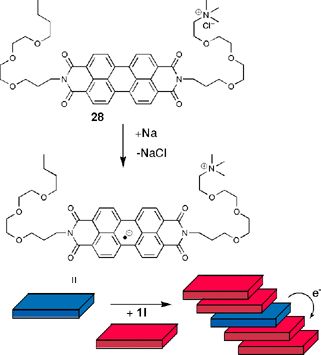 | ||
| Scheme 7 | ||
Multichromophoric dye assemblies
If additional chromophores are attached at the imide groups of the perylene bisimides, energy and electron transfer processes are expected for the photoexcited (super)molecules. Upon self-aggregation of such molecules functional nanostructures are formed which may function as efficient light-harvesting and charge-transporting units for applications in solar cells (videinfra). Recent examples for this approach are compounds 29–31 in Chart 4.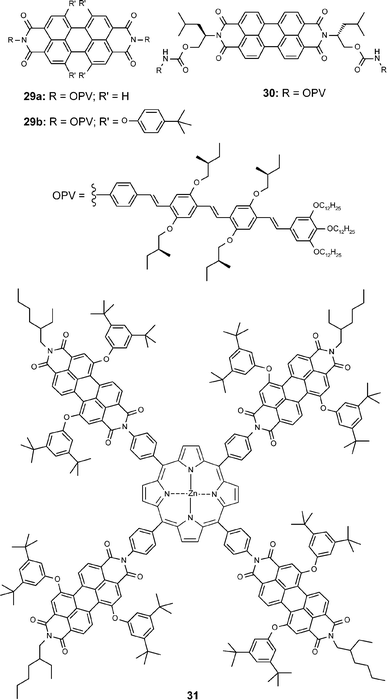 | ||
| Chart 4 | ||
Compounds 29 and 30 are composed of strongly luminescent electron-rich OPV (oligophenylenevinylene) moieties (compounds of this type have been widely employed in electroluminescent devices) and strongly luminescent electron-poor perylene bisimide moieties.65–67 The combination of these dyes in one supermolecule leads to complete quenching of the fluorescence of both dyes due to an efficient electron transfer process in the excited state. Based on the extended aromatic surfaces of these molecules, a pronounced tendency of these dyes towards aggregation was envisioned. UV/Vis dilution studies for the OPV–perylene bisimide system 29b revealed a defined π–π-stacking process in methylcyclohexane with a binding constant which is one order of magnitude higher than those of the corresponding simple perylene bisimide dye 21c.67 For 29a an even higher value was expected but not evident in dilute toluene solution.65 Such strong π–π-stacking was, however, found for the related molecule 30 that shows pronounced changes with well-defined isosbestic points in temperature-dependent UV/Vis studies in tetrachloromethane, toluene, and even chloroform. In contrast to the earlier examples, here, hydrogen-bond formation from the carbamate to the perylene bisimide oxygen provides an additional enthalpic contribution to π–π-stacking and leads to dimeric species instead of extended columnar aggregates.66
For Wasielewski's fourfold perylene bisimide-substituted porphyrin 31 a much higher energy for π–π-stacking was deduced from UV/Vis spectra in chloroform, dimethyl sulfoxide, and pyridine.68 Even at low concentrations of 10−5 M and temperatures as high as 100 °C excitonic coupling is observed between the perylene bisimide units suggesting the presence of aggregates. As shown by light scattering and atomic force microscopy (AFM) studies, extended particles are formed with closely stacked perylene bisimide units (ca. 3.5 Å) and more distant zinc porphyrin units (ca. 7 Å). Upon photoexcitation ZnTPP+–PDI− ion pairs are formed which could be conveniently detected by UV/Vis spectroscopy due to the strong and characteristic PDI− absorption band at 720 nm.
Hydrogen-bond-directed self-assembly
The comparison of the Gibbs free energies for the hydrogen-bonded melamine–imide interaction (Fig. 5) with those determined for π–π-stacking of perylene bisimides reveals that these energies are quite similar, at least in aliphatic, aromatic, and chloroaliphatic solvents. Accordingly, a simple self-assembly protocol for defined objects that is solely based on hydrogen-bonding similar to that of metal–ligand coordination should be ruled out. Instead, at the required concentrations for triple hydrogen-bond formation π–π-stacking will also take place. This leads to a rather interesting situation when ditopic perylene bisimide 32 and ditopic complementary melamine 33 are brought together at identical concentrations in a solvent of low polarity. In methylcyclohexane, where perylene bisimide 32 is only sparingly soluble, in the presence of melamine 33 deeply colored and strongly fluorescent solutions are formed (Scheme 8). Concentration-dependent UV/Vis studies show a distinct transition from molecularly dissolved perylene dyes at <10−6 M to aggregated dyes at 10−4 M. Compared to simple one-dimensional aggregates of 21a–e a much faster growth into extended aggregates is observed upon increasing the concentration, and dynamic light scattering experiments confirmed the existence of nanoparticles already at concentrations as low as 10−4 M. Therefore, this system behaves quite similarly to 31 but with the difference that in the system 32·33 the lateral self-assembly is accomplished by hydrogen-bonding and the vertical self-assembly by π–π-stacking interactions. Once these solutions are evaporated on hydrophobic surfaces, intensely luminescent fibrous aggregates are obtained (Fig. 7).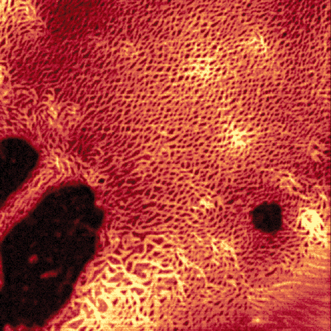 | ||
| Fig. 7 Confocal fluorescence micrograph of mesoscopic superstructures of 32·33 on a silanized glass surface (for details see ref. 45). | ||
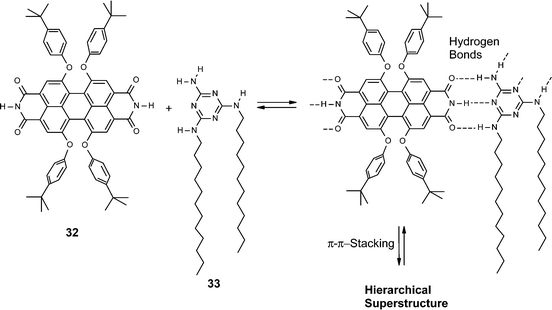 | ||
| Scheme 8 | ||
The influence of π–π-stacking on the fluorescence properties has been studied in solution and also for the mesoscopic assemblies in the solid state. In both cases a significant bathochromic shift and line-broadening were observed compared to the spectra of the isolated chromophores in dilute solution. Once melamine 33 was replaced by melamines 34 bearing chiral side chains derived from amino acids, a similar aggregation process took place and an intense bisignate Cotton effect was observed for the perylene bisimide band. This Cotton effect was explained in terms of a helical stacking of the chromophores that is directed by the chiral side chains of the hydrogen-bonded melamines (Scheme 9).69
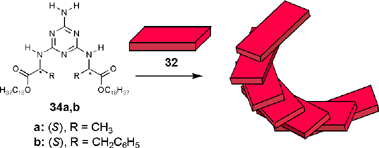 | ||
| Scheme 9 | ||
The same strategy of hydrogen-bond-directed hierarchical growth was successfully applied to synthesize well-defined coaggregates of oligophenylenevinylene (OPV) and perylene bisimide dyes 35·32·35 (Scheme 10).67,70 These assemblies are particularly interesting from the structural as well as a functional point of view. Thus, by means of triple hydrogen-bonding a weak (i.e. reversible) but nonetheless quite rigid bond (three hydrogen-bonds fix the planes of the two molecules to coplanarity) is formed which does not couple the two chromophores electronically. On the other hand, when π–π-stacking of this supramolecular unit in the orthogonal direction takes place, strong coupling between each dye manifold occurs. In the given example this coupling was particularly strong as evidenced by a pronounced bathochromic shift of the perylene bisimide absorption band from 562 nm to 604 nm upon aggregation. Moreover, CD spectroscopy showed not only a significant bisignate Cotton effect for the OPV absorption band pointing at a helical stacking of the OPV dyes but, additionally, a strong CD for the perylene bisimide band, which is indicative of chirality induction from the optically active OPVs to the perylene bisimide backbone. Thus, while in solution the interconversion between the two twisted perylene bisimide conformers (cf.Fig. 2) is fast and both enantiomers are present in equal concentration (racemic mixture), upon aggregation under the influence of chiral OPVs the M-stereoisomer is favored in the diastereomeric coaggregate. Furthermore, the P-helical superhelices and double-helical nanostructures of these coaggregated dyes could be visualized by AFM that revealed even higher hierarchical levels of self-organization.
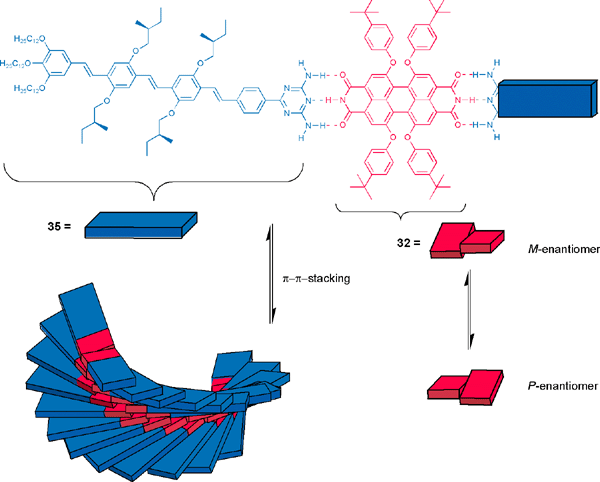 | ||
| Scheme 10 | ||
For the structurally related diazadibenzoperylene dyes, which are accessible from perylene bisimides by reduction of the carbonyl groups and subsequent dehydrogenation, liquid-crystalline mesophases could be induced by coordination of the aromatic aza ligands to tridodecyloxybenzoic acids by hydrogen-bonding.71
Ionic self-assembly
Whilst the supramolecular ordering in the above-mentioned examples was controlled by highly directional noncovalent forces (hydrogen-bonding and π–π-stacking), other forces also proved successful for directing the structural growth over several length scales. In this regard the simple approach of Iverson and Tam-Chang72 and Faul, Antonietti and coworkers73 to organize perylene bisimide dyes by ionic self-assembly seems to be particularly interesting because it is applicable in water. Thus, the rather simple amphiphilic perylene bisimide derivative 36a (Chart 5) could be dissolved in water at rather high concentrations leading to a lyotropic nematic mesophase. This solution could be transferred to a solid support with alignment of the molecules along the spreading direction. After removal of the solvent highly anisotropic films could be obtained whose dichroic ratio rivals commercial polarizers.72 In a more recent communication the same authors have applied this concept to the more extended quaterrylene bisimide dyes affording a long-wavelength polarizing material.74 In the case of 36b, iodide was exchanged with the anionic surfactant dihexadecyl phosphate to afford a thermotropic liquid-crystalline material with a complex phase behavior. This LC was characterized by differential scanning calorimetry (DSC) as well as by wide and small angle X-ray scattering.73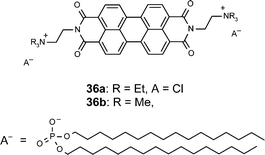 | ||
| Chart 5 | ||
Self-assembly on surfaces and interfaces
Formation of well-ordered organic–inorganic interfaces has a long tradition in the field of organic epitaxy, and perylene tetracarboxylic acid dianhydride (PTCDA; 2) is one of the most studied compounds in this field. This is because deposition of PTCDA onto various substrates under ultrahigh vacuum (UHV) conditions affords almost perfectly ordered overlayers.75 In these layers the molecular arrangement is governed by quadrupolar forces between the PTCDA molecules as well as by intermolecular interactions between the dyes and the substrate. On graphite surfaces the intermolecular interactions prevail leading to a densely packed 2D layer where the quadrupolar interactions between the molecules are optimized in a herringbone arrangement of the dyes (Fig. 8).76 On Ag(111) the packing is quite similar, but owing to a stronger dye–substrate interaction a less dense packing is observed and commensurability of the monolayer and substrate lattice is realized.77 In this particular case, the perylene bisanhydride π-system seems to be bound to the substrate by ‘chemical bonding’ in contrast to the more common physisorption as observed on graphite.78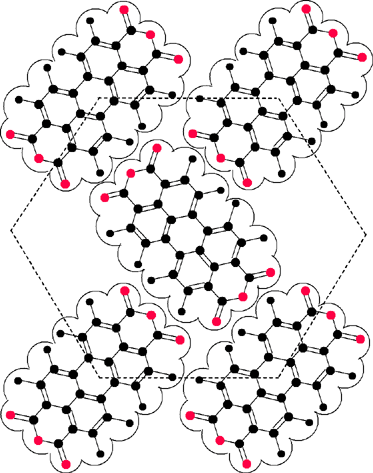 | ||
| Fig. 8 Organization of PTCDA molecules 2 on graphite. The dashed lines visualize the quasi-hexagonal symmetry of the PTCDA monolayer. | ||
Recently, perylene bisimide 1a has also been deposited under UHV conditions and rows of hydrogen-bonded molecules could be nicely observed on the hydrogen-terminated Si(111) surface by scanning tunneling microscopy (STM).79 This study has shown that the strength and directionality of two hydrogen-bonds suffices to overcome quadrupolar forces. If additional melamine (37) is co-adsorbed, a two-dimensional honeycomb network is formed through triple hydrogen-bonding (Fig. 9) similar to the self-assembly process in solution shown in Scheme 8. The large pores of this hexagonal network could be used to accommodate heptameric C60 clusters.80 Such controlled deposition of electronically active molecules, e.g. perylene bisimides and fullerenes, in well-defined surface patterns might be of considerable interest for various applications including molecular electronics.
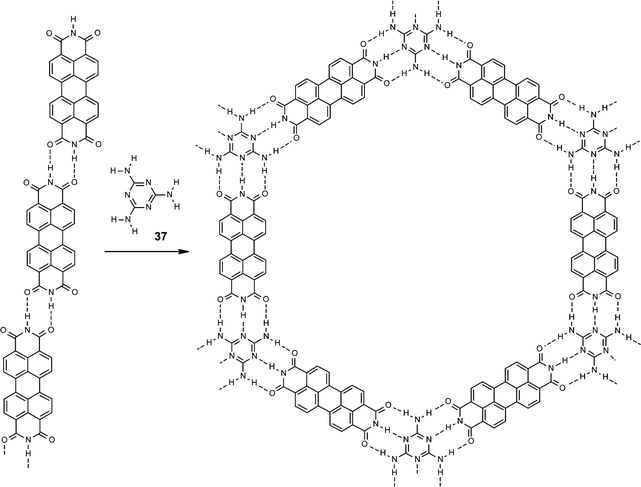 | ||
| Fig. 9 Formation of a hexagonal network of melamine (37)/perylene bisimide (1a) assemblies by triple hydrogen-bonds according to STM studies on Ag/Si(111). | ||
For less volatile perylene bisimides such as OPV–perylene bisimide 29b, two-dimentional organization could be achieved at the graphite (HOPG)/liquid interface of a 1-phenyloctane solution and investigated by STM.81 Remarkably, for this extended π-conjugated molecule, not only could the individual units of the perylene bisimide, the two OPVs, and the alkyl chains be resolved, but, additionally, the two π-conjugated units could be distinguished by bias-dependent tunneling currents. Thus, at negative sample bias a higher tunneling current was observed for the OPV molecular parts, whereas at positive sample bias a higher tunneling current was observed for the perylene bisimide part. This intriguing observation has been attributed to the energetic position of the HOMO (located on OPV) and LUMO (located on perylene bisimide) frontier orbitals with respect to the Fermi level of graphite. Again, it is the nodes in the HOMO and the LUMO of the perylene bisimides that enable such diode-type properties to be observed within a single molecule.
Recently, other approaches to the self-assembly of perylene bisimides by π–π-stacking at interfaces have been reported.82,83 In the first case the earlier discussed perylene bisimides 1k and 1l were compressed at the air/water interface in a Kibron trough to prepare Langmuir–Blodgett films. In the second study, disulfides 38 were self-assembled on plain and nanostructured gold surfaces (Fig. 10). On both types of gold surface, π–π-aggregation of the perylene bisimide dyes takes place as confirmed by fluorescence spectroscopy. However, the luminescence intensity remains high only for the gold nanoparticle-assembled dyes.
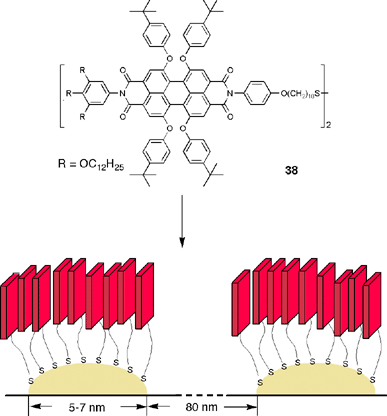 | ||
| Fig. 10 Self-assembly of thiol-functionalized perylene bisimide dyes 38 on a patterned surface of gold nanoparticles. | ||
Conclusions and perspectives
During the last years several new perylene bisimide dyes have become available with a broad range of optical and electrochemical properties. More than for any other class of dyes (maybe with the exception of porphyrins), the supramolecular organization of perylene bisimides by directional forces like hydrogen-bonding and metal–ligand coordination has been developed and diverse architectures such as squares, rosettes, and extended fibrous assemblies have been obtained in solution and at interfaces. In addition, these directional noncovalent forces have proven to be useful to control the π–π-stacking of perylene bisimides.Such π–π-stacking may strongly change the molecular properties of these dyes (absorption, fluorescence) and enable (or improve) desirable properties like exciton and charge transport which are of importance for applications of π-conjugated molecules in organic field effect transistors and light emitting devices as well as in organic solar cells. For all of these applications remarkable progress has recently been achieved with perylene bisimide-based materials, however, yet mostly by rather conventional crystalline derivatives.4,9 Therefore, it remains to be established that rational organization of perylene bisimides in space on the nano- and meso-scopic scale can lead to better technical performance on the device level. For solution- and melt-processable liquid-crystalline perylene bisimides notable charge carrier mobilities and electroluminescence properties have already been realized.3,58,60 However, as in the case of polymer-based electronics and photovoltaics limitations in charge carrier mobilities and exciton diffusion length are directly related to the structural and energetic disorder in not precisely and equidistantly organized dyes. This challenge may be encountered by additional well-positioned supramolecular interaction sites that provide the required set screws for fixation of the functional dyes within nano- and meso-scopic aggregates in time and space.
Besides these goals in the field of advanced technology, appealing structures have become accessible by receptor-functionalized perylene bisimides upon self-assembly with complementary guests. The simplicity of such an approach for the construction of multichromophoric architectures is clearly very attractive since it does not require time-consuming multi-step covalent synthesis, and it can lead to quite large but defined objects. Even larger sized objects of this type are found in the light-harvesting complexes that occur in nature which contain chlorophyll, carotenes and other dyes held together by noncovalent interactions.84 Therefore, it is our hope that the concepts outlined in this feature article for perylene bisimide dyes will be also applicable to organize other kinds of dyes and that eventually a construction kit for supramolecular electronics and photonics85 will become reality.
Acknowledgements
Our part of this reviewed research has been accomplished by many productive coworkers and collaborators I had the pleasure to work with. All individual contributions are acknowledged with gratitude and are specified in the references. Work in the author's laboratory was supported by the Deutsche Forschungsgemeinschaft, Volkswagen Foundation, Fonds der Chemischen Industrie, BASF and Alexander von Humboldt Foundation.References
- H. Zollinger, Color Chemistry, 3rd edn., VCH, Weinheim, 2003 Search PubMed.
- W. Herbst and K. Hunger, Industrial Organic Pigments: Production, Properties, Applications, 2nd edn., WILEY-VCH, Weinheim, 1997 Search PubMed.
- C. W. Struijk, A. B. Sieval, J. E. J. Dakhorst, M. Van Dijk, P. Kimkes, R. B. M. Koehorst, H. Donker, T. J. Schaafsma, S. J. Picken, A. M. van de Craats, J. M. Warman, H. Zuilhof and E. J. R. Sudhölter, J. Am. Chem. Soc., 2000, 122, 11057–11 066 CrossRef.
- C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002, 14, 99–117 CrossRef CAS.
- S. K. Lee, Y. Zu, A. Herrmann, Y. Geerts, K. Müllen and A. J. Bard, J. Am. Chem. Soc., 1999, 121, 3513–3520 CrossRef CAS.
- H. Quante and K. Müllen, Angew. Chem., Int. Ed. Engl., 1995, 34, 1323–1325 CrossRef CAS.
- F. Würthner, Angew. Chem., Int. Ed, 2001, 40, 1037–1039 CrossRef CAS.
- K.-Y. Law, Chem. Rev., 1993, 93, 449–486 CrossRef CAS.
- For recent work, see: L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119–1122 Search PubMed; A. Yakimov and S. R. Forrest, Appl. Phys. Lett., 2002, 80, 1667–1669 CrossRef CAS.
- G. Geissler and H. Remy (Hoechst AG), Ger. Pat. Appl., DE 1130099, 1959 (Chem. Abstr., 1962, 57, P11346f) Search PubMed.
- M. P. O'Neil, M. P. Niemczyk, W. A. Svec, D. Gosztda, G. L. Gaines III and M. R. Wasielewski, Science, 1992, 257, 63–66 CrossRef CAS.
- S. Prathapan, S. I. Yang, J. Seth, M. A. Miller, D. F. Bocian, D. Holten and J. S. Lindsey, J. Phys. Chem. B., 2001, 105, 8237–8248 CrossRef CAS.
- J. M. Serin, D. W. Brousmiche and J. M. J. Frechet, Chem. Commun., 2002, 2605–2607 RSC.
- M. Sadrai and G. R. Bird, Opt. Commun., 1984, 51, 62–64 CrossRef CAS; H.-G. Löhmannsröben and H. Langhals, Appl. Phys. B, 1989, 48, 449–452; R. Reisfeld and G. Seybold, Chimia, 1990, 44, 295–297 CAS.
- W. E. Ford and P. V. Kamat, J. Phys. Chem., 1987, 91, 6373–6380 CrossRef CAS.
- G. Seybold and G. Wagenblast, Dyes Pigm., 1989, 11, 303–317 CrossRef; G. Seybold and A. Stange (BASF AG), Ger. Pat., DE 35 45 004, 1987 (Chem. Abstr., 1988, 108, 77134c) Search PubMed.
- R. Gvishi, R. Reisfeld and Z. Burshtein, Chem. Phys. Lett., 1993, 213, 338–344 CrossRef CAS.
- S. Mais, J. Tittel, T. Basché, C. Bräuchle, W. Göhde, H. Fuchs, G. Müller and K. Müllen, J. Phys. Chem. A, 1997, 101, 8435–8440 CrossRef CAS; H. Langhals, H. Jaschke, U. Ring and P. von Unold, Angew. Chem., Int. Ed., 1999, 38, 201–203 CrossRef CAS.
- T. Weil, U. M. Wiesler, A. Herrmann, R. Bauer, J. Hofkens, F. C. De Schryver and K. Müllen, J. Am. Chem. Soc., 2001, 123, 8101–8108 CrossRef CAS; T. Weil, E. Reuther and K. Müllen, Angew. Chem., Int. Ed., 2002, 41, 1900–1904 CrossRef CAS; R. Gronheid, J. Hofkens, F. Köhn, T. Weil, E. Reuther, K. Müllen and F. C. De Schryver, J. Am. Chem. Soc., 2002, 124, 2418–2419 CrossRef CAS; G. De Belder, G. Schweitzer, S. Jordens, M. Lor, S. Mitra, J. Hofkens, S. De Feyter, M. Van der Auweraer, A. Herrmann, T. Weil, K. Müllen and F. C. De Schryver, ChemPhysChem, 2001, 1, 49–55 CrossRef.
- L. Zang, R. Liu, M. W. Holman, K. T. Nguyen and D. M. Adams, J. Am. Chem. Soc., 2002, 124, 10640–10641 CrossRef CAS.
- H. Langhals, Heterocycles, 1995, 40, 477–500 CrossRef CAS and refs. cited therein.
- H. Langhals, S. Demmig and H. Huber, Spectrochim. Acta, 1988, 44A, 1189–1193 CrossRef.
- A. Stange, G. Wagenblast and G. Seybold, BMFT-Bericht T 86–216, Fachinformationszentrum Karlsruhe, Germany, 1986 Search PubMed.
- R. Iden and G. Seybold (BASF AG), Ger. Pat. Appl., DE 3434059 A1, 1985 (Chem. Abstr., 1985, 103, 38696q) Search PubMed.
- M. Sandrai, L. Hadel, R. R. Sauers, S. Husain, K. Krogh-Jespersen, J. D. Westbrook and G. R. Bird, J. Phys. Chem., 1992, 96, 7988–7996 CrossRef.
- A. Böhm, H. Arms, G. Henning and P. Blaschka, (BASF AG), Ger. Pat. Appl., DE 19547209 A1, 1997 (Chem. Abstr., 1997, 127, 96569g) Search PubMed.
- U. Rohr, P. Schlichting, A. Böhm, M. Groß, K. Meerholz, C. Bräuchle and K. Müllen, Angew. Chem., Int. Ed., 1998, 37, 1434–1437 CrossRef CAS; U. Rohr, C. Kohl, K. Müllen, A. van de Craats and J. Warman, J. Mater. Chem., 2001, 11, 1789–1799 RSC.
- F. Würthner, Z. Chen and V. Stepanenko, unpublished results.
- A. Böhm, H. Arms, G. Henning and P. Blaschka (BASF AG), Ger. Pat. Appl., DE 19547210 A1, 1997 (Chem. Abstr., 1997, 127, 96570a) Search PubMed.
- M. J. Ahrens, M. J. Fuller and M. R. Wasielewski, Chem. Mater., 2003, 15, 2684–2686 CrossRef CAS.
- Y. Zhao and M. R. Wasielewski, Tetrahedron Lett., 1999, 40, 7047–7050 CrossRef CAS; A. S. Lukas, Y. Zhao, S. E. Miller and M. R. Wasielewski, J. Phys. Chem. B, 2002, 106, 1299–1306 CrossRef CAS.
- H. Langhals, S. Demmig and T. Potrawa, J. Prakt. Chem., 1991, 333, 733–748 CrossRef CAS.
- H. Langhals, J. Karolin and L. B.-A. Johansson, J. Chem. Soc., Faraday Trans., 1998, 94, 2919–2922 RSC.
- A. Rademacher, S. Märkle and H. Langhals, Chem. Ber., 1982, 115, 2927–2934 CrossRef CAS.
- F. Würthner, C. Thalacker, S. Diele and C. Tschierske, Chem. Eur. J., 2001, 7, 2245–2253 CrossRef CAS; F. Würthner and C. Thalacker (BASF AG), Ger. Pat. Appl., DE 10039232 A1, 2000 (Chem. Abstr., 2002, 136, 185323) Search PubMed.
- J. Salbeck, H. Kunkely, H. Langhals, R. W. Saalfrank and J. Daub, Chimia, 1989, 43, 6–9 CAS.
- Z. Chen, M. G. Debije, T. Debaerdemaeker, P. Osswald and F. Würthner, ChemPhysChem, 2004, 5, 137–140 CrossRef CAS.
- F. Graser and E. Hädicke, Liebigs Ann. Chem., 1980, 1994–2011 CAS; F. Graser and E. Hädicke, Liebigs Ann. Chem., 1984, 483–494 CAS; E. Hädicke and F. Graser, Acta Crystallogr., Sect. C, 1986, 42, 189–195 CrossRef; E. Hädicke and F. Graser, Acta Crystallogr., Sect. C, 1986, 42, 195–198 CrossRef; G. Klebe, F. Graser, E. Hädicke and J. Berndt, Acta Crystallogr., Sect. B, 1989, 45, 69–77 CrossRef.
- P. Zugenmaier, J. Duff and T. L. Bluhm, Cryst. Res. Technol., 2000, 35, 1095–1115 CrossRef CAS.
- F. Würthner, A. Sautter and J. Schilling, J. Org. Chem., 2002, 67, 3037–3044 CrossRef.
- S. Hien, PhD Thesis, University of Regensburg, Germany, 1995.
- J. Hofkens, T. Vosch, M. Maus, F. Köhn, M. Cotlet, T. Weil, A. Herrmann, K. Müllen and F. C. De Schryver, Chem. Phys. Lett., 2001, 333, 255–263 CrossRef CAS.
- P. M. Kazmaier and R. Hoffmann, J. Am. Chem. Soc., 1994, 116, 9684–9691 CrossRef CAS.
- F. Würthner, Habilitation Thesis, University Ulm, Germany, 2001. Upon request, a pdf file of the summary of this thesis (51 pages) is available from the author.
- F. Würthner, C. Thalacker and A. Sautter, Adv. Mater., 1999, 11, 754–758 CrossRef CAS; F. Würthner, C. Thalacker, A. Sautter, W. Schärtl, W. Ibach and O. Hollricher, Chem. Eur. J., 2000, 6, 3871–3886 CrossRef CAS.
- R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2003, 5–16 RSC.
- E. E. Neuteboom, E. H. A. Beckers, S. C. J. Meskers, E. W. Meijer and R. A. J. Janssen, Org. Biomol. Chem., 2003, 1, 198–203 RSC.
- F. Würthner, A. Sautter, D. Schmid and P. J. A. Weber, Chem. Eur. J., 2001, 7, 894–902 CrossRef CAS.
- F. Würthner and A. Sautter, Org. Biomol. Chem., 2003, 1, 240–243 RSC.
- C.-C. You and F. Würthner, J. Am. Chem. Soc., 2003, 125, 9716–9725 CrossRef CAS.
- R. Dobrawa, D. G. Kurth and F. Würthner, Polymer Preprints, 2004, 45(1), 378–379 Search PubMed; R. Dobrawa and F. Würthner, Chem. Commun., 2002, 1878–1879 RSC.
- W. Wang, L.-S. Li, G. Helms, H.-H. Zhou and A. D. Q. Li, J. Am. Chem. Soc., 2003, 125, 1120–1121 CrossRef CAS; A. D. Q. Li, W. Wang and L.-Q. Wang, Chem. Eur. J., 2003, 9, 4594–4601 CrossRef CAS.
- E. E. Neuteboom, S. C. J. Meskers, E. W. Meijer and R. A. J. Janssen, Macromol. Chem. Phys., 2004, in press Search PubMed.
- W. Wang, J. J. Han, L.-Q. Wang, L.-S. Li, W. J. Shaw and A. D. Q. Li, Nano Lett., 2003, 3, 455–458 CrossRef CAS.
- W. Wang, W. Wan, H.-H. Zhou, S. Q. Niu and A. D. Q. Li, J. Am. Chem. Soc., 2003, 125, 5248–5249 CrossRef CAS.
- R. A. Cormier and B. A. Gregg, J. Phys. Chem. B, 1997, 101, 11004–11006 CrossRef CAS; R. A. Cormier and B. A. Gregg, Chem. Mater., 1998, 10, 1309–1319 CrossRef CAS.
- C. Göltner, D. Pressner, K. Müllen and H. W. Spieß, Angew. Chem., Int. Ed. Engl., 1993, 32, 1660–1662 CrossRef.
- T. Hassheider, S. A. Benning, H.-S. Kitzerow, M.-F. Achard and H. Bock, Angew. Chem., Int. Ed., 2001, 40, 2060–2063 CrossRef CAS.
- S. Benning, H.-S. Kitzerow, H. Bock and M.-F. Achard, Liq. Cryst., 2000, 27, 901–906 CrossRef CAS.
- S. Alibert-Fouet, S. Dardel, H. Bock, M. Oukachmih, S. Archambeau, I. Seguy, P. Jolinat and P. Destruel, ChemPhysChem, 2003, 4, 983–985 CrossRef.
- M. G. Debije, Z. Chen and F. Würthner, unpublished results.
- S. Ferrere, A. Zaban and B. A. Gregg, J. Phys. Chem. B, 1997, 101, 4490–4493 CrossRef CAS.
- B. A. Gregg, J. Sprague and M. W. Peterson, J. Phys. Chem. B, 1997, 101, 5362–5369 CrossRef CAS.
- B. A. Gregg and R. A. Cormier, J. Am. Chem. Soc., 2001, 123, 7959–7960 CrossRef CAS.
- A. Syamakumari, A. P. H. J. Schenning and E. W. Meijer, Chem. Eur. J., 2002, 8, 3353–3361 CrossRef CAS.
- E. Peeters, P. A. van Hal, S. C. J. Meskers, R. A. J. Janssen and E. W. Meijer, Chem. Eur. J., 2002, 8, 4470–4474 CrossRef CAS.
- F. Würthner, Z. Chen, P. Osswald, C.-C. You, P. Jonkheijm, J. v. Herrikhuyzen, F. Hoeben, A. P. H. J. Schenning, E. W. Meijer, E. H. A. Beckers, S. C. J. Meskers and R. A. J. Janssen, unpublished results.
- T. van der Boom, R. T. Hayes, Y. Zhao, P. J. Bushard, E. A. Weiss and M. R. Wasielewski, J. Am. Chem. Soc., 2002, 124, 9582–9590 CrossRef.
- C. Thalacker and F. Würthner, Adv. Funct. Mater, 2002, 12, 209–218 CrossRef CAS.
- A. P. H. J. Schenning, J. v. Herrikhuyzen, P. Jonkheijm, Z. Chen, F. Würthner and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 10252–10253 CrossRef CAS.
- A. Sautter, C. Thalacker and F. Würthner, Angew. Chem., Int. Ed., 2001, 40, 4425–4428 CrossRef CAS.
- I. K. Iverson and S.-W. Tam-Chang, J. Am. Chem. Soc., 1999, 121, 5801–5802 CrossRef CAS.
- Y. Guan, Y. Zakrevskyy, J. Stumpe, M. Antonietti and C. F. J. Faul, Chem. Commun., 2003, 894–895 RSC.
- S.-W. Tam-Chang, W. Seo, I. K. Iverson and S. M. Casey, Angew. Chem., Int. Ed., 2003, 42, 897–900 CrossRef CAS.
- E. Umbach, K. Glöckler and M. Sokolowski, Surf. Sci., 1998, 402–404, 20–31 CrossRef CAS.
- C. Ludwig, B. Gompf, W. Glatz, J. Petersen, W. Eisenmenger, M. Möbius, U. Zimmermann and N. Karl, Z. Phys. B, 1992, 86, 397–404 CAS.
- K. Glöckler, C. Seidel, A. Soukopp, M. Sokolowski, E. Umbach, M. Böhringer, R. Berndt and W.-D. Schneider, Surf. Sci., 1998, 405, 1–20 CrossRef CAS.
- M. Eremtchenko, J. A. Schaefer and F. S. Tautz, Nature, 2003, 425, 602–605 CrossRef CAS.
- C. Ludwig, B. Gompf, J. Petersen, R. Strohmaier and W. Eisenmenger, Z. Phys. B, 1994, 93, 365–373 CAS; B. Uder, C. Ludwig, J. Petersen, B. Gompf and W. Eisenmenger, Z. Phys. B., 1995, 97, 389–390 CAS.
- J. A. Theobald, N. S. Oxtoby, M. A. Phillips, N. R. Champness and P. H. Beton, Nature, 2003, 424, 1029–1031 CrossRef CAS.
- A. Miura, Z. Chen, H. Uji-i, S. De Feyter, M. Zdanowska, P. Jonkheijm, A. P. H. J. Schenning, E. W. Meijer, F. Würthner and F. C. De Schryver, J. Am. Chem. Soc., 2003, 125, 14968–14969 CrossRef CAS.
- G. Sui, J. Orbulescu, M. Mabrouki, R. M. Leblanc, S. Liu and B. A. Gregg, ChemPhysChem, 2002, 1041–1044 CrossRef CAS.
- U. Haas, C. Thalacker, J. Adams, J. Fuhrmann, S. Riethmüller, U. Beginn, U. Ziener, M. Möller, R. Dobrawa and F. Würthner, J. Mater. Chem., 2003, 13, 767–772 RSC.
- G. McDermott, S. M. Prince, A. A. Freer, A. M. Hawthornthwaite-Lawless, M. Z. Papiz, R. J. Cogdell and N. W. Isaacs, Nature, 1995, 374, 517–521 CrossRef CAS.
- V. Balzani and F. Scandola, Supramolecular Photochemistry, Ellis Horwood Ltd., Chichester, 1991 Search PubMed.
| This journal is © The Royal Society of Chemistry 2004 |