Design, synthesis and evaluation of bifunctional inhibitors of type II dehydroquinase†
Miguel D.
Toscano
a,
Martyn
Frederickson
a,
David P.
Evans
a,
John R.
Coggins
b,
Chris
Abell
*a and
Concepción
González-Bello
c
aUniversity Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW
bInstitute of Biomedical and Life Sciences, Division of Biochemistry and Molecular Biology, University of Glasgow, Glasgow, UK G12 8QQ
cDepartamento de Química Orgánica y Unidad Asociada al C.S.I.C., Facultad de Química, Universidad de Santiago de Compostela, 15782 Santiago de Compostela, Spain
First published on 15th May 2003
Abstract
Inhibitors of type II dehydroquinase were designed to straddle the two distinct binding sites identified for the inhibitor (1S,3R,4R)-1,3,4-trihydroxy-5-cyclohexene-1-carboxylic acid and a glycerol molecule in a crystallographic study of the Streptomyces coelicolor enzyme. A number of compounds were designed to incorporate characteristics of both ligands. These analogues were synthesized from quinic acid, and were assayed against type I (Salmonella typhi) and type II (S. coelicolor) dehydroquinases. None of the analogues showed inhibition for type I dehydroquinase. Six of the analogues were shown to have inhibition constants in the micromolar to low millimolar range against the S. coelicolor type II dehydroquinase, while two showed no inhibition. The binding modes of the analogues in the active site of the S. coelicolor enzyme were studied by molecular docking with GOLD1.2. These studies suggest a binding mode where the ring is in a similar position to (1S,3R,4R)-1,3,4-trihydroxy-5-cyclohexene-1-carboxylic acid in the crystal structure and the side-chain occupies part of the glycerol binding-pocket.
Introduction
Dehydroquinase (EC 4.2.1.10, 3-dehydroquinate dehydratase) catalyses the conversion of 3-dehydroquinate (1) into 3-dehydroshikimate (2) (Scheme 1).1 This is the third step in the shikimate pathway leading to chorismate, the precursor to the aromatic amino-acids L-phenylalanine, L-tryptophan and L-tyrosine, and other key metabolic intermediates (e.g.p-aminobenzoic acid).2 The pathway is present in plants, fungi, bacteria and some parasites, and inhibitors of shikimate pathway enzymes are candidates for antibiotic, anti-parasitic and herbicidal agents (e.g. the herbicide glyphosate inhibits the sixth enzyme, 5-enolpyruvyl shikimate-3-phosphate (EPSP) synthase).3 The conversion of 3-dehydroquinate into 3-dehydroshikimate is also a step on the quinate pathway in fungi.4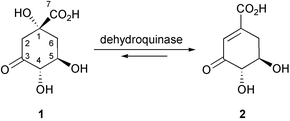 | ||
| Scheme 1 The reaction catalysed by dehydroquinase. | ||
There are two structurally distinct forms (type I and type II) of dehydroquinase, which catalyse the same transformation by distinct mechanisms.1 The type I enzyme catalyses the syn elimination of water from 3-dehydroquinate via imine intermediates that are covalently attached to a conserved lysine side chain.5 The type II enzyme catalyses an anti elimination via an enol or enolate intermediate.6 Inhibitors have been designed which specifically inhibit type I7 and type II dehydroquinases.8
The crystal structures of both type I and type II dehydroquinases have been reported.9 Recently, the structure of the Streptomyces coelicolor type II dehydroquinase has been determined at 1.8 Å resolution, with the potent reversible inhibitor 3 bound at the active site (PDB code: 1GU1, the crystals were obtained at pH 8.5 in Tris buffer in the presence of PEG8Kcrystallisation Na/K phosphate and sodium tartrate).10 This complex identifies a number of key interactions involved in inhibitor binding, and sheds light on aspects of the catalytic mechanism of the enzyme. Also present in this structure was a molecule of glycerol bound 3.7 Å away from the inhibitor (Fig. 1). The glycerol originated from the enzyme storage buffer, but its adventitious appearance in the crystal structure suggests additional binding interactions which can be exploited in inhibitor design.
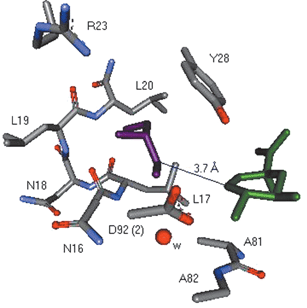 | ||
| Fig. 1 Selected view of the active-site of type II dehydroquinase (S. coelicolor) from the crystal structure (1.8 Å resolution, PDB code 1GU1) showing the relative position of 3 (green) and the glycerol molecule (purple). The distance between the two closest carbon atoms in 3 and glycerol is 3.7 Å. | ||
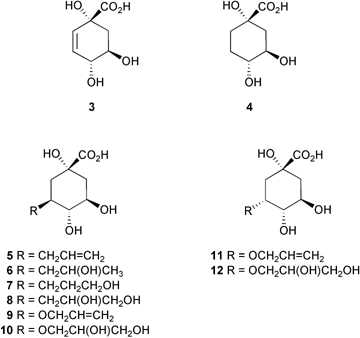
There are several examples of inhibitor design where two inhibitors which bind to distinct regions of an enzyme active site have been incorporated into a larger inhibitor with significant increases in potency.11 In this paper we explore this idea by making compounds 5–12 which straddle the binding sites of both 3 and the glycerol molecule, and incorporate structural features from both. The results of inhibition studies with these compounds against type I (Salmonella typhi) and type II dehydroquinase (S. coelicolor), and of molecular docking studies into the active-site of S. coelicolor type II dehydroquinase using GOLD1.212 are described.
Results
Synthesis
All of the target compounds were accessible from quinic acid (13). The first series 5–8 involved replacement of the C-3 hydroxy with an allyl group, which was subsequently elaborated to introduce hydroxy groups. The second series 11–12 involved attachment and modification of an allyl group attached to the C-3 hydroxy group of quinic acid (13). A similar approach using compounds epimeric at C-3 was used to make 9–10.The strategy used for making 5–8 (Scheme 2) involved the initial preparation of the protected trans-diaxial allylbenzoate analogue (14). This was made in 3 steps and 47% yield, following the methodology of Widlanski et al.13 The synthesis of 5 was achieved by a two step deprotection of 14. The lactone 14 was treated with a catalytic amount of sodium methoxide in methanol to remove the benzoate group and form the methyl ester 15 (87%). This was purified by extraction into water, washing with diethyl ether to remove the methyl benzoate, followed by lyophilization. Conversion of 15 to the 3-allyl quinic acid analogue 5 was achieved in 90% yield by ester hydrolysis, followed by treatment with Dowex 50 (H+) ion-exchange resin and lyophilization. When deprotection of 14 was attempted in a single step using sodium hydroxide, the product was obtained in a mixture with benzoic acid which proved difficult to separate even by ion-exchange chromatography.
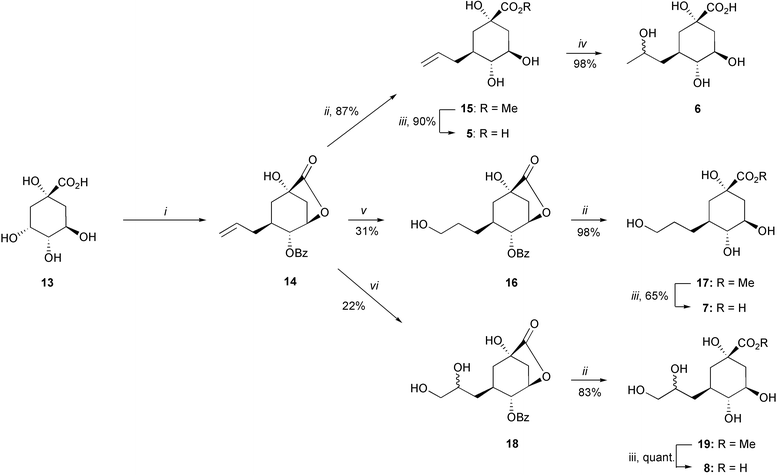 | ||
| Scheme 2 Reagents and conditions: (i) ref. 13; (ii) MeOH, NaOMe, RT; (iii) 1. NaOH, H2O, RT, 2. Dowex 50 (H+); (iv) HCl, H2O, Δ; (v) 1. BH3–THF, THF, 0 °C, 2. NaBO3, H2O, 0 °C; (vi) OsO4 (cat), NMO, acetone–H2O (1 : 1), RT. | ||
The introduction of the 2′-hydroxy group into the side chain olefin in 15 to form 6 was achieved in 98% yield by acid-catalysed hydroxylation with concentrated hydrochloric acid under reflux for 24 hours. The product was obtained as an approximately 1 : 1 mixture of diastereoisomers, epimeric at C-2′. The two diastereoisomers could be partially resolved by HPLC using a preparative organic acids column, but it was not possible to separate them cleanly. The analogue 6 could also be formed quantitatively by stirring 5 in concentrated hydrochloric acid for 4 days at room-temperature.
The anti-Markovnikov mono-hydroxylated compound 16 was obtained in 31% yield by hydroboration of 14, using borane–THF complex followed by work-up with sodium perborate and water. The modest yield was due to the difficult purification of the product, which is partially deprotected during the work-up. Compound 16 was purified by column chromatography on silica gel and deprotected by treatment with sodium methoxide in methanol to give the methyl ester 17 (98%) which was then hydrolysed to the target compound 7 (65%). The hydroxylation reaction was also attempted using borane–dimethyl sulfoxide complex but a lower yield was obtained. Furthermore the standard work-up with the hydrogen peroxide–sodium hydroxide totally deprotected the hydroxy groups around the ring.
The first step towards the dihydroxylated analogue 8 required catalytic osmylation of 14. This was achieved using N-methylmorpholine N-oxide to reoxidise the catalyst14 to form 18 as a 1 : 1 mixture of diastereoisomers (epimeric at C-2′) in 22% yield. Attempts to separate the diastereoisomers by HPLC on an organic acids column and by acetylation and benzoylation of the free hydroxy groups were unsuccessful. The deprotection of 18 was carried out by initial treatment with sodium methoxide in methanol, to give the methyl ester 19 (83%). This was then hydrolysed with sodium hydroxide, followed by ion exchange to yield the acid 8 quantitatively. Osmylation of 14 using trimethylamine N-oxide as a reoxidant and dichloromethane as the only solvent15 led to no reaction. Catalytic osmylation on the methyl ester 15 using N-methylmorpholine N-oxide was not successful.
Compounds 9 and 10 were synthesized from the protected alcohol 20 prepared using a previously reported protocol (Scheme 3).16 Ketone 21 was obtained via oxidation of 20 with pyridinium dichromate under dry conditions (77%), and was subsequently reduced to give alcohol 22.17 Allylation of 22 was achieved by treatment with allyl methyl carbonate and a catalytic amount of Pd2(dba)2/dppb to give 23 in 69% yield.18 This step was particularly difficult because a competitive diallylation process was observed, which could be overcome by adding the solution of the carbonate slowly to the reaction mixture. The desired acid 9 was obtained by a two-step deprotection sequence. First, treatment with aqueous trifluoroacetic acid cleaved the bismethoxyacetal protecting group giving 24 in 94% yield, which upon basic hydrolysis followed by ion exchange gave the acid 9 (97%). Treatment of the allyloxy ether 23 with osmium tetraoxide (catalytic) resulted in hydroxylation with concomitant lactonization to afford 25 in 78% yield. Deprotection of 25 was achieved in the same manner as for 23, to give the acid 10 as a 1.2 : 1 mixture of diastereoisomers in 95% yield.
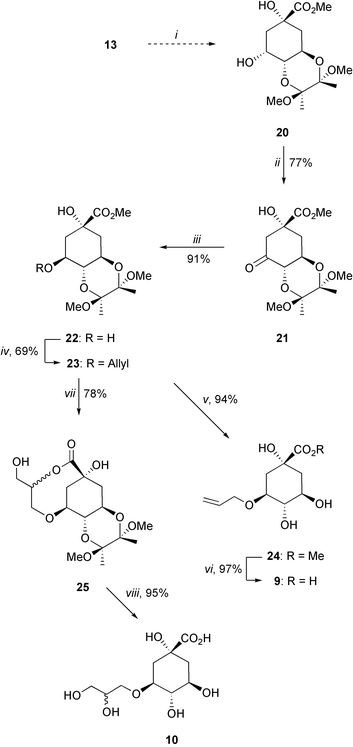 | ||
Scheme 3
Reagents and conditions: (i) ref. 16; (ii) PDC, 4 Å MS, DCM, RT; (iii) ref. 17; (iv) CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2OCO2Me, Pd2(dba)2, dppb, THF, Δ; (v) TFA–H2O (20 : 1), RT; (vi) 1. LiOH, H2O, RT, 2. Amberlite IR-120; (vii) OsO4 cat, NMO, dioxane–H2O, RT; (viii) 1. TFA–H2O (20 : 1), RT, 2. LiOH, H2O, RT, 3. Amberlite IR-120. CHCH2OCO2Me, Pd2(dba)2, dppb, THF, Δ; (v) TFA–H2O (20 : 1), RT; (vi) 1. LiOH, H2O, RT, 2. Amberlite IR-120; (vii) OsO4 cat, NMO, dioxane–H2O, RT; (viii) 1. TFA–H2O (20 : 1), RT, 2. LiOH, H2O, RT, 3. Amberlite IR-120. | ||
Compounds 11 and 12 were synthesized from the protected alcohol 20 (Scheme 4). The allyl side-chain was introduced using palladium coupling with allyl methyl carbonate in quantitative yield to give 26. Treatment of 3-allyloxy derivative 26 with aqueous trifluoroacetic acid led to deprotection of the bis-methoxyacetal together with partial allyl migration to afford a chromatographically separable mixture of 3-allyloxy and 4-allyloxy derivatives 27 (72%) and 28 (25%). Finally, hydrolysis of the methyl ester 27 under basic conditions followed by ion exchange gave acid 11 in 89% yield. Hydroxylation of 26 with catalytic osmium tetraoxide gave 29 in 76% yield, which was deprotected using a combination of TFA, lithium hydroxide and Amberlite IR-120 to give 12 as a 1.2 : 1 mixture of diastereoisomers.
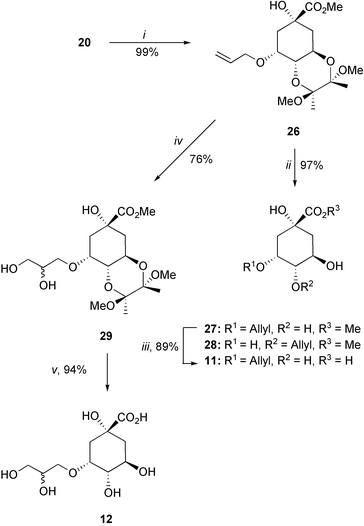 | ||
| Scheme 4
Reagents and conditions: (i) CH2CHCH2OCO2Me, Pd2(dba)2, dppb, THF, Δ; (ii) TFA–H2O (20 : 1); (iii) 1. LiOH, H2O, RT, 2. Amberlite IR-120; (iv) OsO4(cat), NMO, dioxane–H2O, RT; (v) 1. TFA–H2O (20 : 1), RT, 2. LiOH, H2O, RT, 3. Amberlite IR-120. | ||
Assay results
Inhibition studies against type I dehydroquinase (S. typhi) and type II dehydroquinase (S. coelicolor) were performed with compounds 5, 7, 9 and 11, and compounds 6, 8, 10 and 12 as the prepared diastereomeric mixtures. The UV spectrophotometric assay6c was used to measure the initial rate of product formation, detecting the enone-carboxylate chromophore at 234 nm in 3-dehydroshikimate (2). The Ki values (Table 1) were obtained from Dixon plots (1/vvs. [I]).19| Compound | Type I dehydroquinase S. typhia | Type II dehydroquinase S. coelicolora |
|---|---|---|
| a K M values of 16 µM (type I dehydroquinase, S. typhi) and 250 µM (type II dehydroquinase, S. coelicolor) were obtained at the assay conditions. b Ref. 8. | ||
| 3 | 3000 ± 1000b | 30 ± 10b |
| 4 | 4500 ± 500b | 600 ± 200b |
| 5 | >20000 | 420 ± 50 |
| 6 (R + S) | >20000 | >20000 |
| 7 | >20000 | 180 ± 20 |
| 8 (R + S) | >20000 | 3000 ± 500 |
| 9 | >20000 | 1200 ± 150 |
| 10 (R + S) | >20000 | 530 ± 50 |
| 11 | >20000 | >20000 |
| 12 (R + S) | >20000 | 3500 ± 400 |
The most striking observation is that none of the compounds 5–12 showed any measurable inhibition against the type I dehydroquinase. This result was not too surprising as substrate analogues where the carbonyl oxygen in 3-dehydroquinate (1) was replaced by either CH2 or NOH had previously been shown not to inhibit type I dehydroquinase.8
The best inhibitor of type II dehydroquinase was 7 which had a Ki of 180 µM. This compared with a KM for substrate of 250 µM under the same assay conditions. The related allyl compound 5 lacking the terminal hydroxy group had an increased Ki of 420 µM. A similar pattern was observed for the dihydroxylated ether 10 (Ki 530 µM) and corresponding allyl compound 9 (Ki 1200 µM). The diols 8 and 12 showed modest inhibition (3.0 and 3.5 mM, respectively), while no inhibition was observed for the secondary alcohol 6 and the C-3 epimeric allyl ether 11.
Molecular modelling
The structures (ligands) of 4, 5, 7, 9, 11 and each stereoisomer of 6, 8, 10 and 12 were docked in the active-site of the structure of type II dehydroquinase from S. coelicolor (receptor) (Fig. 1), using the program GOLD (version 1.2).12 The structures of the ligands were prepared using the program SYBYL6.520 and energy minimised using the Tripos force field. The receptor was also prepared using SYBYL6.5. The structures of 3, the glycerol molecule, and all of the water molecules (except a key structural water molecule that is present in all the crystal structures, shown in Fig. 1) were removed from the crystal structure. No energy minimisation was performed on the receptor since the crystal structure used has the enzyme in the desired conformation, with bound ligands. All the ligands were docked as carboxylate anions and 25 independent GOLD runs were performed for each ligand.Ligand 3 was initially docked as a control, and the result compared to the crystal structure of the enzyme inhibitor complex. All 25 GOLD runs gave results within 0.5 Å of each other, and the position of the docked ligand in the active-site coincided with the position in the crystal structure to within 0.3 Å RMSD. The reduced analogue 4 also docked consistently at the same site with an RMSD of 1.0 Å.
The docking of ligands 5–9 also gave highly consistent results. All 25 GOLD runs for each ligand showed the 6-membered ring in a similar position to 3, with only small variations in the position of the side-chains. Compounds 10, 11 and 12 also docked in this position in the large majority of the 25 experiments, but other binding modes were also found where the ring position was substantially changed. These were not considered further in this study.
Fig. 2 shows the docking results of ligands 7, 8R and 10S, compared with the position of 3 and the glycerol molecule in the crystal structure. All the ligands have the ring in a similar position as 3 and the side-chain in the glycerol binding-pocket. For ligands 5–10 the ring adopts a chair conformation. However, in the dockings of ligands 11 and 12 the ring is flipped into a boat-conformation. This moves the side-chain onto an equatorial position so that it extends into the glycerol binding-pocket, and avoids steric clashes with protein side chains below the ring. From the ligand positions in the docking results no H-bond interactions are recognised between the hydroxy groups in the ligand side-chains and the enzyme.
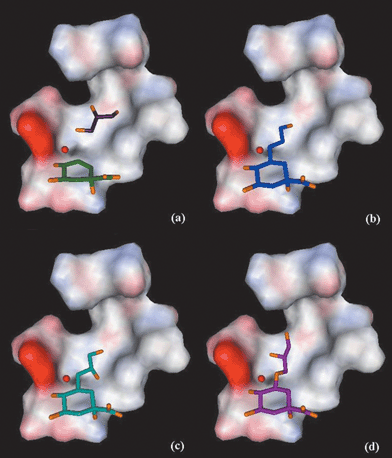 | ||
| Fig. 2 Results from the docking experiments with GOLD1.2: positions of the ligands in the active-site of type II dehydroquinase (S. coelicolor), against a surface (coloured by electrostatic potential)21 calculated for the protein residues shown in Fig. 1, omitting Y28 for clarity. Oxygen atoms are shown in orange. (a) 3 (green) and glycerol (purple), (b) 7 (blue), (c) 8R (light blue) and (d) 10S (magenta). | ||
Discussion and conclusions
The observation of a glycerol molecule adventitiously bound at a distal site in the structure of the complex between S. coelicolor type II dehydroquinase and inhibitor 3 inspired the design of bifunctional inhibitors which would straddle the two sites and incorporate features of both 3 and glycerol. The target compounds 5–12 were all synthesized from quinic acid (13). Compounds with secondary hydroxy groups in their side chain were prepared and tested as diastereomeric mixtures.None of the compounds 5–12 inhibited type I dehydroquinase. This was expected as the type I enzyme mechanism involves initial attack on the C-3 carbonyl of 3-dehydroquinate (1) by an active site lysine.5a Substrate analogues substituted at C-3 would be expected to suffer unfavourable steric clashes with this side chain.
Five compounds showed competitive inhibition against type II dehydroquinase, although all were less potent than the original inhibitor 3. The most potent inhibitor 7 has a Ki below the KM for substrate, and is more potent than 4, the reduced analogue of 3 with sp3 hybridisation at C-3. In analysing the inhibition data several trends were discerned:
(i) Introduction of a hydroxy group in the side chain three atoms remote from C-3 of quinic acid 13 is favorable. This is most clearly seen in the comparison of 7vs. 5.
(ii) Extending the side chain to four atoms appears to be less favourable, e.g.5vs. 9 and 7vs10.
(iii) Introduction of a hydroxy group in the side chain two atoms remote from C-3 is unfavourable. This is clearly seen in the comparison of 6 with 5 and 7, and also 8vs. 7.
(iv) There is a preference for the side chain to be on the β-face, i.e. the (R)- stereochemistry at C-3 is less favourable when comparing 10vs. 12 and 9vs. 11.
These trends consistently account for the relative affinities of the compounds 5–12. They are further supported by consideration of the structures of the ligands docked into the active site using GOLD, shown in Fig. 2.
Fig. 2a shows the position of 3 and the glycerol molecule in the active-site. Fig. 2b overlays of the docked structure of 7, where the side-chain reaches into the glycerol binding-pocket and the terminal hydroxy overlaps the C-1 position of the glycerol molecule. Docking of 5 shows the ligand in a similar position to 7.
The docking of the dihydroxy ligand 8R is shown on Fig. 2c. This suggests a position where the secondary hydroxy group at C-2′ abuts Leu17. This result is independent of whether or not the adjacent water molecule is removed. Ligands 6R, 6S and 8S dock in a similar position to 8R. The terminal primary hydroxy group on C-3′ of 8 reaches into the glycerol pocket and may account for its affinity relative to 6. The extended diol 10S is shown in Fig. 2d. The diol 10 has the longest side chain and it docks so as to overlap both C-1 and C-2 of glycerol. This might have been expected to increase the affinity compared to 7, but must be offset against the increased entropic cost of immobilizing the longer side chain. Finally, the docking results for ligands 11 and 12 (both with (R)-stereochemistry at C-3) suggest they bind in a similar position to 10S. However, to accommodate the side-chain in the glycerol pocket the six-membered ring is flipped into a boat conformation. This conformational switch, which would be forced by steric crowding below C-3 (see Fig. 1) preventing the side chain extending downwards, is likely to disrupt hydrogen bonding to the hydroxys at C-4 and C-5, contributing to their reduced affinity.
In conclusion, the strategy of making inhibitors combining structural features of both 3 and glycerol has resulted in the formation of new inhibitors. The docking experiments suggest that most of the compounds bind in a similar manner to 3, with their side chains extending into the glycerol binding site. The modest affinity of the inhibitors is probably a consequence of the entropic cost of immobilising a flexible side chain, and the sensitivity of binding to sp2 hybridisation at C-2 and C-3 of the six membered ring. Both of these issues are being addressed in the design of the next generation of inhibitors.
Experimental
General
All organic solvents were freshly distilled prior to use and Milli-Q deionised water was used for all biochemical work. Analytical thin layer chromatography was carried out on commercial silica gel 60 0.25 mm plates using either UV absorption, iodine staining, ceric molybdate solution or potassium manganate(VII) spray for visualisation. RF values are quoted with respect to the solvent system used to develop the plate. Column chromatography was carried out using 230–400 mesh silica gel 60. Melting points are uncorrected. NMR spectra were recorded in deuterated solvents. In the spectra of diastereomeric mixtures, all resolvable peaks are given. Infrared spectra were recorded as NaCl plates, Nujol mulls or KBr discs. [α]D values are given in 10−1 deg cm2 g−1. Ultraviolet–visible spectra were recorded using black-walled quartz cuvettes. Carboxylic acids were analysed or purified by HPLC on a preparative (300 mm × 16 mm) Bio-Rad Aminex Ion Exclusion HPX-87H Organic Acids column. The eluant used for these columns was 0.05 M aqueous formic acid, at a flow rate of 1.0 ml min−1. All procedures involving the use of ion-exchange resins were carried out at room temperature and used Milli-Q deionised water. Dowex 50W-X8 (H) (cation exchanger) and Amberlite IR-120 (H) (cation exchanger) were washed alternately with water, 10% HCl, water, 10% sodium hydroxide, water, 10% HCl and finally with water before use. Purified S. typhi type I dehydroquinase and S. coelicolor type II dehydroquinase were stored in aliquots as concentrated solutions in glycerol–water at −20 °C and 4 °C, respectively.Dehydroquinase assay
The dehydroquinase enzymes were assayed by monitoring the increase in absorbance at 234 nm in the UV spectrum due to the absorbance of the enone-carboxylate chromophore of 3-dehydroshikimate (2ε = 1.2 × 104 M−1cm−1). The assays were performed at 25 °C in potassium phosphate (0.05 M, pH 7) buffer for type I dehydroquinase and Tris-HCl (0.05 M, pH 7) buffer for type II dehydroquinase. The assay mixture was prepared in situ and the assay was initiated by addition of the enzyme solution to the mixture. Solutions of 3-dehydroquinate (1) were calibrated by equilibration with type I dehydroquinase and measurement from the change in UV absorbance at 234 nm. In these assays the concentration of inhibitor (I) was varied for a constant concentration of substrate and enzyme. The assays were repeated for a number of different concentrations of substrate and the Ki values were obtained through a Dixon plot (1/vvs. [I]).GOLD docking
All ligands and the receptor were prepared using SYBYL6.5 and used as MOL2 files. The ligands were designed as carboxylate anions and their structure was energy minimised using the Tripos force-field prior to docking. Each ligand was docked using GOLD1.2 in 25 independent genetic algorithm (GA) runs, and for each of these a maximum number of 100000 GA operations was performed on a single population of 50 individuals. Operator weights for crossover, mutation and migration in the entry box were used as default parameters (95, 95 and 10, respectively), as well as the hydrogen bonding (4.0 Å) and van der Waals (2.5 Å) parameters. The position of the active-site was introduced and the radius was set to 10 Å, with the automatic active-site detection on. The “flip ring corners” flag was switched on, while all the other flags were off.Methyl (1S,3R,4R,5S)-5-allyl-1,3,4-trihydroxycyclohexane-1-carboxylate (15)
The allyl carbolactone 1413 (140 mg, 0.47 mmol) was dissolved in methanol (10 ml) and stirred in the presence of sodium methoxide (5 mg) for 24 hours at room temperature. The solvent was removed at reduced pressure, the crude product was taken in water (10 ml) and washed with diethyl ether (3 × 20 ml). The ester15 was lyophilised and collected as an amorphous yellow solid (94 mg, 87%). δH (400 MHz, D2O): 5.65 (1 H, m), 4.95 (2 H, m), 3.62 (3 H, s), 3.60 (1 H, ddd, J = 12.0, 9.4 and 5.1 Hz), 3.05 (1 H, t, J = 9.4 Hz), 2.35 (1 H, dddd, J = 14.2, 6.1, 3.2 and 1.5 Hz), 2.00–1.80 (2 H, m), 1.75–1.60 (3 H, m), 1.45 (1 H, t, J = 14.2 Hz); δC (100 MHz, DEPT, D2O): 175.2 (C), 134.3 (CH), 115.1 (CH2), 75.3 (CH), 72.6 (C), 68.7 (CH), 51.2 (OCH3), 37.8 (CH2), 35.3 (CH2), 34.9 (CH), 33.2 (CH2); HRMS calcd for C11H18O5Na: MNa+, 253.1052. Found: MNa+, 253.1072.(1S,3R,4R,5S)-5-Allyl-1,3,4-trihydroxycyclohexane-1-carboxylic acid (5)
The methyl ester 15 (94 mg, 0.41 mmol) was dissolved in water (10 ml) and taken to pH∼12 with 10% NaOH, and stirred at room temperature overnight. The solution was acidified with Dowex 50 (H+) ion-exchange resin, filtered and lyophilised to give the allyl acid5 as an amorphous white solid (80 mg, 90%). HPLC retention time (organic acids column): 37 minutes; υmax (Nujol)/cm−1 3368b (OH) and 1712s (CO); δH (500 MHz, D2O): 5.80 (1 H, m), 5.05 (2 H, m), 3.70 (1 H, ddd, J = 14.0, 9.3 and 4.9 Hz), 3.17 (1 H, dd, J = 10.1 and 9.3 Hz), 2.45 (1 H, dddd, J = 14.0, 6.3, 3.2 and 1.6 Hz), 2.00 (2 H, m), 1.80 (3 H, m), 1.58 (1 H, dd, J = 14.0 and 12.7 Hz); δC (100 MHz, DEPT, D2O): 179.3 (C), 136.2 (CH), 116.8 (CH2), 77.2 (CH), 74.4 (C), 70.7 (CH), 39.8 (CH2), 37.2 (CH2), 36.9 (CH), 34.0 (CH2); HRMS calcd for C10H16O5: M+ − H, 215.0920. Found: M+ − H, 215.0939.(1S,3R,4R,5S)-5-(2′-Hydroxypropyl)-1,3,4-trihydroxycyclohexane-1-carboxylic acid (6)
The acid 5 (11 mg, 0.052 mmol) was stirred with concentrated hydrochloric acid (2 ml) under reflux for 24 hours. The acidic solution was diluted in water and the solvent and some of the acid removed at reduced pressure. The residue was redissolved in water (5 ml) and lyophilised to give a 1 : 1 mixture of diastereoisomers of the secondary hydroxy acid6 as an amorphous white solid (12 mg, 98%). HPLC retention time (organic acids column): 36 and 37 minutes; υmax (Nujol)/cm−1 3381b (OH) and 1719s (CO); δH (500 MHz, D2O): 4.27 (1 H, tq, J = 9.1 and 6.2 Hz), 3.81 (1 H, ddd, J = 11.0, 9.6 and 4.8 Hz), 3.30 (1 H, dd, J = 10.3 and 9.6 Hz), 2.20 (1 H, ddd, J = 12.0, 6.2 and 6.0 Hz), 1.98 (2 H, m), 1.85 (1 H, dd, J = 13.5 and 3.3 Hz), 1.75 (1 H, dd, J = 13.7 and 11.0 Hz), 1.69 (1 H, t, J = 13.5 Hz), 1.34 (1 H, td, J = 12.0 and 9.1 Hz), 1.25 (3 H, d, J = 6.2 Hz); δC (100 MHz, DEPT, D2O): 179.7 (C), 86.3 (CH), 78.0 (CH), 77.3 (C), 70.4 (CH), 42.0 (CH2), 40.2 (CH), 39.3 (CH2), 37.5 (CH2), 22.4 (CH3); only ten peaks observed in 13C spectrum of the mixture. HRMS calcd for C10H15O5: M( − H+ − H2O), 215.0920. Found: M( − H+ − H2O), 215.0937.(1S,3S,4R,5S)-5-(3′-Hydroxypropyl)-4-benzoyl-1-hydroxycyclohexane-1,3-carbolactone (16)
A solution of borane–THF complex (1.89 ml ca. 1.0 M in THF, 1.89 mmol, 2 equiv.) was added to a solution of the allyl carbolactone 14 (286 mg, 0.94 mmol) in THF (5 ml), at 0 °C in an ice-bath. The solution was stirred for 1 hour, and a solution of sodium perborate (286 mg, 1.89 mmol) in water (5 ml) was added and the ice-bath removed. The solution was stirred for a further 2 hour at room temperature. The THF was removed at reduced pressure, the product was dissolved in ethyl acetate (20 ml) and washed with water (3 × 20 ml). The organic layer was dried with anhydrous Na2SO4 and the solvent removed at reduced pressure. The crude product was purified by column chromatography on silica gel, eluting with ethyl acetate–petroleum ether 40–60 (3 : 1). The carbolactone16 (94 mg, 31%) was obtained as a colourless oil. RF 0.22 [EtOAc–petroleum ether 40–60 (3 : 1)]; υmax (NaCl)/cm−1 3452b (OH) and 1790s (CO), 1723s (CO) and 1601s (Ar); δH (400 MHz, CDCl3): 8.10 (2 H, d, J = 8.0 Hz), 7.60 (1 H, t, J = 8.0 Hz), 7.46 (2 H, t, J = 8.0 Hz), 5.20 (1 H, dd, J = 5.0 and 2.3 Hz), 4.89 (1 H, t, J = 5.0 Hz), 3.63 (2 H, m), 3.52 (1 H, s), 2.58–2.45 (2 H, m), 2.33 (1 H, dd, J = 13.3 and 9.1 Hz), 2.23 (1 H, m), 1.86 (1 H, m), 1.75–1.60 (3 H, m), 1.50 (1 H, m); δC (100 MHz, CDCl3): 181.2, 167.5, 135.8, 131.8, 131.5, 130.8, 77.8, 74.2, 72.9, 64.4, 39.5, 39.3, 39.2, 33.0, 32.9; HRMS calcd for C17H20O6Na: MNa+, 343.1143. Found: MNa+, 343.1158.Methyl (1S,3R,4R,5S)-5-(3′-hydroxypropyl)-1,3,4-trihydroxycyclohexane-1-carboxylate (17)
The lactone 16 (87 mg, 0.27 mmol) was dissolved in methanol (5 ml) and stirred in the presence of sodium methoxide (5 mg) for 2 hours at room temperature. The solvent was removed at reduced pressure, the crude product was taken in water (10 ml) and washed with diethyl ether (3 × 10 ml). The product was lyophilised to give ester17 as a colourless glass (66 mg, 98%). υmax (Nujol)/cm−1 3330b (OH) and 1730b (OH); δH (400 MHz, D2O): 3.66 (3 H, s), 3.60 (1 H, m), 3.50 (2 H, t, J = 6.5 Hz), 3.08 (1 H, td, J = 9.0 and 7.2 Hz), 1.99 (1 H, ddd, J = 13.2, 4.0 and 3.1 Hz), 1.85−1.45 (6 H, m), 1.36 (1 H, m), 1.15 (1 H, m); δC (100 MHz, DEPT, D2O): 178.7 (C), 79.6 (CH), 76.0 (C), 73.0 (CH), 63.3 (CH2), 54.6 (OMe), 42.3 (CH2), 39.7 (CH2), 38.3 (CH), 29.7 (CH2), 28.4 (CH2).(1S,3R,4R,5S)-5-(3′-Hydroxypropyl)-1,3,4-trihydroxycyclohexane-1-carboxylic acid (7)
The methyl ester 17 (10 mg, 0.040 mmol) was dissolved in water (2 ml), the pH adjusted to 12 with sodium hydroxide (10%) and the solution stirred at room temperature for 2 hours. The solution was acidified with Amberlite IR-120 (H+) ion-exchange resin, filtered and lyophilised to give the 3′-hydroxy acid7 as a colourless glass (6 mg, 65%); HPLC retention time (organic acids column): 28 minutes; υmax (Nujol)/cm−1 3393b (OH) and 1711s (CO); δH (400 MHz, D2O): 3.66 (1 H, ddd, J = 11.8, 9.5 and 4.6 Hz), 3.54 (2 H, t, J = 6.5 Hz), 3.12 (1 H, t, J = 9.5 Hz), 2.02 (1 H, ddd, J = 13.3, 4.6 and 3.1 Hz), 1.83–1.52 (6 H, m), 1.41 (1 H, ddd, J = 17.1, 11.2 and 6.5 Hz), 1.20 (1 H, m); δC (100 MHz, DEPT, D2O): 179.4 (C), 77.6 (CH), 74.2 (C), 70.4 (CH), 61.7 (CH2), 39.7 (CH2), 37.0 (CH2), 36.6 (CH), 27.8 (CH2), 26.5 (CH2); HRMS calcd for C10H18O6Na: MNa+, 257.1001. Found: MNa+, 257.1001.(1S,3R,4R,5S)-5-(2′,3′-Dihydroxypropyl)-4-benzoyl-1-hydroxycyclohexane-1,3-carbolactone (18)
To a solution of the carbolactone 14 (167 mg, 0.55 mmol) in acetone (2 ml) was added N-methylmorpholine oxide (76.7 mg, 0.65 mmol), water (2 ml) and osmium tetraoxide (1.1 mg, 55 µl of 2.5% wt. solution in tert-butanol, 5.5 µM). The mixture was degassed with nitrogen and stirred for 20 hours under argon, with additional osmium tetraoxide (1.1 mg, 55 µl of 2.5% solution in tert-butanol, 5.5 µM) added after 7 hours. A saturated solution of sodium bisulfite (10 ml) was added to destroy excess of osmium tetraoxide. The acetone was removed at reduced pressure and the product was extracted with ethyl acetate (2 × 20 ml). The extract was washed with water (4 × 20 ml). The solution was dried with MgSO4 and evaporated at reduced pressure. The product was purified by column chromatography on silica gel eluting with ethyl acetate to give the diol (1 ∶ 1 mixture of diastereomers) 18 as a colourless oil (40 mg, 22%). RF 0.25 (ethyl acetate); δH (400 MHz, CDCl3): 7.95 (2 H, d, J = 8.2 Hz), 7.55 (1 H, t, J = 8.2 Hz), 7.40 (2 H, t, J = 8.2 Hz), 5.10 (1 H, m), 4.82 (1 H, t, J = 4.5 Hz), 4.05–3.35 (5 H, m), 2.60–2.20 (4 H, m), 1.94 (1 H, dd, J = 10.2 and 9.0 Hz), 1.65 (1 H, m), 1.45 (1 H, m); δC (100 MHz, CDCl3): 179.9, 179.8, 166.1, 166.0, 134.1, 134.0, 130.1 (2×), 129.7, 129.6, 129.0, 128.9, 76.1, 76. 0, 72.5, 72.2, 71.0 (2×), 70.5, 70.1, 67.0, 66.8, 38.4, 38.3, 37.8, 37.5, 37.2, 35.9, 34.2, 33.5; HRMS calcd for C17H20O7Na: MNa+, 359.1109. Found: MNa+, 359.1158.Methyl (1S,3R,4R,5S)-5-(2′,3′-dihydroxypropyl)-1,3,4-trihydroxycyclohexane-1-carboxylate (19)
The dihydroxy lactone 18 (40 mg, 0.12 mmol) was dissolved in methanol (10 ml) and stirred in the presence of sodium methoxide (1 mg) for 6 hours at room temperature. The solvent was removed at reduced pressure, the crude product was taken in water (10 ml) and washed with diethyl ether (3 × 20 ml). The solution was lyophilised to give the ester (1 ∶ 1 mixture of diastereomers) 19 as a brown solid (27 mg, 83%). δH (400 MHz, D2O): 3.68 (3 H, s), 3.67 (2 H, m) 3.48 (1 H, ddd, J = 17.9, 12.1 and 4.0 Hz), 3.36 (1 H, ddd, J = 17.9, 12.1 and 7.2 Hz), 3.08 (1 H, td, J = 9.0, 6.3 Hz), 2.02 (1 H, m), 1.90–1.73 (4 H, m), 1.60 (1 H, t, J = 12.0 Hz), 1.22 (1 H, m); δC (100 MHz, DEPT, D2O): 179.5 (2 × C), 81.1 (CH), 80.4 (CH), 76.9 (CH2), 76.8 (CH2), 73.1 (CH), 72.9 (CH), 71.4 (2 × CH), 68.6 (CH2), 67.7 (CH2), 55.0 (2 × OCH3), 42.0 (2 × CH2), 40.8 (CH2), 39.8 (CH2), 37.3 (CH2), 37.2 (CH), 36.9 (CH2), 36.2 (CH).(1S,3R,4R,5S)-5-(2′,3′-Dihydroxypropyl)-1,3,4-trihydroxycyclohexane-1-carboxylic acid (8)
The ester 19 (27 mg, 0.10 mmol) was dissolved in water (10 ml), the pH of the solution adjusted to 12 with sodium hydroxide (10%) and stirred at room temperature overnight. The solution was acidified with Dowex 50 (H+) ion-exchange resin, filtered and lyophilised to give the two diastereisomers of the dihydroxy acid8 (1 ∶ 1 mixture) as a white solid (25 mg, quantitative). HPLC retention time (organic acids column): 22 and 24 minutes; υmax (Nujol)/cm−1 3401b (OH) and 1718s (CO); δH (400 MHz, D2O): 3.73 (1 H, ddd, J = 14.0, 6.5 and 3.4 Hz), 3.64 (1 H, ddd, J = 14.1, 9.2 and 4.5 Hz), 3.53 (1 H, dd, J = 13.0 and 3.4 Hz), 3.36 (1 H, dd, J = 13.0 and 6.5 Hz), 3.10 (1 H, t, J = 9.2 Hz), 1.98 (1 H, ddd, J = 14.0, 4.5 and 3.1 Hz), 1.90–1.65 (4 H, m), 1.60 (1 H, t, J = 12.9 Hz), 1.20 (1 H, m); δC (100 MHz, DEPT, D2O): 179.7 (C), 179.6 (C), 78.0 (CH), 77.8 (CH), 74.3 (CH2), 74.2 (CH2), 70.5 (CH), 70.4 (CH), 70.3 (CH), 68.6 (CH), 65.9 (CH2), 64.9 (CH2), 39.6 (2 × CH2), 38.3 (CH2), 37.3 (CH2), 34.7 (CH), 34.6 (CH2), 34.3 (CH2), 33.7 (CH); HRMS calcd for C10H17O7: M − H, 249.0974. Found: M − H, 249.0968.Methyl (1S,3S,4S,6S,9R)-9-hydroxy-3,4-dimethoxy-3,4-dimethyl-7-oxo-2,5-dioxabicyclo[4.4.0]decane-9-carboxylate (21)
To a stirred suspension of the alcohol 2016 (2.69 g, 8.41 mmol) and activated molecular sieves 4 Å (3.4 g) in dry dichloromethane (80 ml) was added pyridinium dichromate (4.74 g, 12.61 mmol). The resultant suspension was stirred at room temperature for 2 hours and then diluted with diethyl ether (60 ml) and filtered over Celite. The solution was evaporated and the crude reaction was purified by flash chromatography, eluting with ethyl acetate, and then recrystallised from diethyl ether to afford the ketone 21 as white needles (2.06 g, 77%). Mp 194–195 °C (diethyl ether); δH (250 MHz, CDCl3): 4.19 (1 H, dd, J = 10.3 and 0.9 Hz), 4.02 (1 H, td, J = 11.0 and 4.2 Hz), 3.60 (3 H, s), 3.02 (3 H, s), 2.99 (3 H, s), 2.69 (1 H, d, J = 14.3 Hz), 2.28 (1 H, dd, J = 14.3 and 2.8 Hz), 2.12 (1 H, t, J = 12.4 Hz), 1.87 (1 H, m), 1.15 (3 H, s), 1.06 (3 H, s); δC (63 MHz, DEPT, CDCl3): 199.5 (C), 174.1 (C), 100.5 (C), 99.6 (C), 77.2 (CH), 74.0 (C), 67.0 (CH), 53.6 (OCH3), 49.0 (CH2), 48.3 (OCH3), 48.0 (OCH3), 37.8 (CH2), 17.6 (CH3), 17.5 (CH3). MS (CI) m/z (%) 287 [MH+ − CH3OH]; HRMS calcd. For C13H19O7: MH+, 287.1130. Found MH+, 287.1127.Methyl (1S,3S,4S,6S,7S,9R)-7-allyloxy-9-hydroxy-3,4-dimethoxy-3,4-dimethyl-2,5-dioxabicyclo[4.4.0]dec-9-anecarboxylate (23)
To a stirred solution of Pd2(dba)2 (16 mg, 17 µmol) and dppb (29 mg, 68 µmol) in dry tetrahydrofuran (3 ml) under argon was added the alcohol22 (217 mg, 0.68 mmol) and then a solution of allyl methyl carbonate in tetrahydrofuran (1.6 ml, 0.5 M). The resultant green suspension was heated under reflux for 20 hours. The solvent was removed under reduced pressure and the crude mixture was purified by flash chromatography eluting with diethyl ether–hexane (75%) to afford the allyl ether23 as a colourless oil (170 mg, 69%). [α]20D +136 (c 1.40 in CHCl3); υmax (NaCl)/cm−1 3700b (OH) and 1733s (CO); δH (250 MHz, CDCl3): 5.91–5.76 (1 H, m), 5.24–5.06 (2 H, m), 3.92–3.75 (4 H, m), 3.67 (3 H, s), 3.40 (1 H, t, J = 9.6 Hz), 3.22 (3 H, s), 3.18 (3 H, s), 2.32 (1 H, ddd, J = 13.9, 4.6 and 2.8 Hz), 2.14 (1 H, ddd, J = 13.9, 4.2 and 2.8 Hz), 1.85–1.64 (2 H, m), 1.27 (3 H, s), 1.23 (3 H, s); δC (63 MHz,DEPT, CDCl3): 172.5 (C), 134.0 (CH), 117.0 (CH3), 99.5 (C), 99.5 (C), 79.0 (C), 76.2 (CH), 66.5 (CH), 65.7 (CH3), 64.7 (CH), 52.3 (OCH3), 47.8 (2 × OCH3), 37.3 (CH2), 34.9 (CH2), 17.6 (2 × CH3); MS (CI) m/z (%) 329 [(MH+) − HOCH3]; HRMS calcd for C16H25O7: MH+, 329.1600. Found: MH+, 329.1601.Methyl (1S,3R,4R,5R)-3-allyloxy-1,4,5-trihydroxycyclohexane-1-carboxylate (24)
A solution of acetal 23 (117 mg, 0.33 mmol) in 2 ml of a solution of 20 : 1 (v/v) of trifluoroacetic acid–water was stirred for 30 min and then the solvent was concentrated in vacuo. The crude mixture was purified by flash chromatography eluting with ethyl acetate–dichloromethane–methanol (7 : 2 : 1) to afford 24 as a colourless oil (78 mg, 94%). [α]20D −0.2 (c 2.6 in H2O); υmax (NaCl)/cm−1 3399b (OH) and 1731s (CO); δH (300 MHz, D2O): 6.01–5.98 (1 H, m), 5.46–5.32 (2 H, m), 4.08 (2 H, d, J = 5.9 Hz), 3.88 (3 H, s), 3.85 (1 H, m), 3.41 (1 H, t, J = 9.1 Hz), 2.42 (2 H, dd, J = 2.8 and 11.9 Hz), 1.94 (2 H, dd, J = 12.3 and 13.5 Hz); δC (125 MHz, DEPT, D2O): 174.0 (C), 133.2 (CH), 118.2 (CH2), 79.1 (C), 78.7 (CH), 68.6 (2 × CH), 65.8 (CH2), 52.9 (CH3), 37.1 (2 × CH2); MS (CI) m/z (%) 247 (MH+); HRMS calcd for C11H19O6: MH+, 247.1174. Found: MH+, 247.1182.(1S,3R,4R,5R)-3-Allyloxy-1,4,5-trihydroxycyclohexane-1-carboxylic acid (9)
A solution of the methyl ester 24 (70 mg, 0.28 mmol) in aqueous lithium hydroxide (3 ml, 0.2M) was stirred for 1 hour. The reaction mixture was diluted with water and was washed with diethyl ether (3 × 5 ml). The aqueous layer was treated with Amberlite IR-120 until the pH was 6.0. The resin was filtered and washed with water. The filtrate was freeze-dried to afford the acid9 as a colourless oil (67 mg, 97%). [α]20D −0.5 (c 2.2 in H2O); υmax (NaCl)/cm−1 3399b (OH) and 1683s (CO); δH (300 MHz, D2O): 6.11–6.00 (1H, m), 5.46–5.30 (2H,m), 4.05 (2 H, d, J = 5.9 Hz), 3.85–3.76 (2 H, m), 3.41 (1 H, t, J = 9.2 Hz), 2.39 (2 H, dd, J = 4.1 and 12.3 Hz), 1.88 (2 H, t, J = 12.8 Hz); δC (125 MHz, DEPT, D2O): 177.3 (C), 133.8 (CH), 117.8 (CH2), 79.9 (C), 79.0 (CH), 69.0 (2 × CH), 65.6 (CH2), 37.6 (2 × CH2); MS (CI) m/z (%) 233 (MH+); HRMS calcd for C10H17O6: MH+, 233.1025. Found: MH+, 233.1018.Methyl (1S,3S,4S,6S,7S,9R)-7-(2′,3′-dihydroxy)propyloxy-9-hydroxy-3,4-dimethoxy-3,4-dimethyl-2,5-dioxabicyclo[4.4.0]decane-9-carboxylate (25)
To a stirred solution of allyl ether 23 (87 mg, 0.24 mmol) and N-methylmorpholine oxide (34 mg, 0.29 mmol) in 50% aqueous dioxane (4 ml) was added a freshly made aqueous solution of sodium tetraoxide (0.3 ml, 0.12 M). After stirring for 1 hour ethyl acetate was added and then saturated sodium bisulfite. The reaction mixture was stirred for 20 min. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (3 × 10 ml). The combined organic layers were dried (anhydrous Na2SO4) and concentrated in vacuo. The crude residue was purified by flash chromatography eluting with 95% ethyl acetate–methanol to afford a 1.2 : 1 mixture of diastereoisomers of the lactone25 (68 mg, 78%) as white needles. υmax (NaCl)/cm−1 3425b (OH) and 1750s (CO); δH (250 MHz, CDCl3): 4.58 (m, 1H), 3.94–3.66 (6 H, m), 3.48 (1 H, t, J = 9.6 Hz), 3.27 (3 H, s), 3.23 (3 H, s), 2.64 (br s), 2.45–2.16 (2 H, m), 2.08–1.77 (2 H, m), 1.30 (3 H, s), 1.28 (3 H, s); δC (63 MHz, DEPT, CDCl3): 170.2 (C), 99.7 (C), 99.6 (C), 77.4 (C), 77.3 (C), 76.0 (CH), 66.4 (CH), 64.6 (CH), 61.4 (CH2), 61.3 (CH2), 59.2 (CH2), 59.1 (CH2), 48.0 (OCH3), 47.9 (OCH3), 39.4 (CH2), 34.7 (CH2), 17.7 (CH3), 17.6 (CH3); MS (CI) m/z (%) 331 [(MH+) − HOCH3]; HRMS calcd for C15H23O8: MH+, 331.1393. Found: MH+, 331.1393.(1S,3R,4R,5R)-1,4,5-Trihydroxy-3-(2′,3′-dihydroxy)propyloxycyclohexane-1-carboxylic acid (10)
A solution of acetal 25 (46 mg, 0.13 mmol) in 2 ml of a solution of trifluoroacetic acid–water (20 : 1 (v/v)) was stirred for 30 min and then concentrated in vacuo. The crude residue was redissolved in an aqueous lithium hydroxide (1.7 ml, 0.2 M) and stirred for 1 hour. The aqueous layer was washed with diethyl ether (3 × 5 ml) and then was treated with Amberlite IR-120 until the pH was 6.0. The resin was filtered and washed with water. The filtrate was freeze-dried to afford a 1.2 : 1 mixture of diastereoisomers of the acid10 as a colourless oil (34 mg, 95%). υmax (Nujol)/cm−1 3390b (OH) and 1682s (CO); δH (300 MHz, D2O): 3.70 (1 H, br s), 3.55–3.20 (5 H, m), 3.13 (1 H, t, J = 9.2 Hz), 2.10 (2 H, d, J = 11.5 Hz), 1.60 (2 H, t, J = 12.5 Hz); δC (75 MHz, DEPT, D2O): 166.1 (C), 165.6 (C), 82.0 (C + CH), 73.6 (CH), 72.0 (2 × CH), 68.0 (CH2), 65.6 (CH2), 40.5 (CH2), 40.3 (CH2); MS (CI) m/z (%) 249 [(MH+) − H2O]; HRMS calcd for C10H17O7: MH+, 249.0974. Found: MH+, 249.0970.Methyl (1S,3S,4S,6S,7R,9R)-7-allyloxy-9-hydroxy-3,4-dimethoxy-3,4-dimethyl-2,5-dioxabicyclo[4.4.0]decane-9-carboxylate (26)
To a stirred solution of Pd2(dba)2 (14 mg, 15.8 µmol) and dppb (27 mg, 63 µmol) in dry tetrahydrofuran (2 ml) under argon was added the alcohol 20 (200 mg, 0.63 mmol) and then a solution of allyl methyl carbonate (85 µl, 0.75 mmol) in dry tetrahydrofuran (1.5 ml). The resultant green suspension was heated at 60 °C for 2 h. The solvent was removed under reduced pressure and the crude mixture was purified by flash chromatography eluting with 75% diethyl ether–hexane to afford the allyl ether26 as a colourless oil (226 mg, 99%). [α]20D +125 (c 1.9 in CHCl3); υmax (NaCl)/cm−1 3479b (OH) and 1737s (CO); δH (300 MHz, CDCl3): 5.80 (1 H, m), 5.11 (2 H, m), 4.22 (2 H, m), 4.07–3.86 (2 H, m), 3.64 (3 H, s), 3.45 (1 H, ddd, J = 18.2, 10.2 and 2.5 Hz), 3.14 (3 H, s), 3.13 (3 H, s), 2.25 (2 H, m), 1.90–1.74 (2 H, m), 1.19 (3 H, s, CH3), 1.16 (3 H, s, CH3); δC (75 MHz, DEPT, CDCl3): 173.4 (C), 134.3 (CH), 116.8 (CH2), 99.6 (C), 99.1 (C), 83.9 (C), 75.8 (CH), 73.2 (CH), 72.1 (CH2), 62.5 (CH), 52.3 (OCH3), 47.6 (OCH3), 47.5 (OCH3), 39.0 (CH2), 36.6 (CH2), 17.5 (CH3), 17.4 (CH3); MS (CI) m/z (%) 329 [(MH+) − HOCH3]; HRMS calcd for C16H25O7: MH+, 329.1600. Found: MH+, 329.1591.Methyl (1S,3S,4R,5R)-3-allyloxy-1,4,5-trihydroxycyclohexane-1-carboxylate (27) and methyl (1S,3S,4R,5R)-4-allyloxy-1,3,5-trihydroxycyclohexane-1-carboxylate (28)
A solution of the acetal 26 (174 mg, 0.48 mmol) in a mixture 20 : 1 (v/v) of trifluoroacetic acid–water was stirred at room temperature for 15 min. The solvent was removed under reduced pressure and the crude mixture was purified by flash chromatography eluting with 50% acetone–dichloromethane to afford the 3-allyloxy ester27 (85 mg, 72%) and the 4-allyloxy ester28 (30 mg, 25%), both as colourless oils.(1S,3S,4R,5R)-3-Allyloxy-1,4,5-trihydroxycyclohexane-1-carboxylic acid (11)
A solution of the ester 27 (34 mg, 0.14 mmol) in aqueous lithium hydroxide (1.7 ml, 0.2 M) was stirred at room temperature for 2 hours. The resultant solution was diluted with water and treated with Amberlite IR-120 until the pH was 6.0. The resin was filtered and washed with water. The filtrate was concentrated to afford the acid11 (29 mg, 89%) as a colourless oil. [α]20D −25 (c 1.7 in H2O); υmax (NaCl)/cm−1 3418b (OH) and 1713s (CO); δH (300 MHz, D2O): 6.02 (1 H, m), 5.40 (1 H, d, J = 17.4 Hz), 5.31 (1 H, d, J = 10.2 Hz), 4.26–4.07 (3 H, m), 3.71 (1 H, dd, J = 11.2 and 2.6 Hz), 2.24–1.97 (4 H m); δC (75 MHz, DEPT, D2O): 176.1 (C), 131.2 (CH), 115.4 (CH2), 73.7 (CH), 73.0 (C), 70.9 (CH), 67.9 (CH2), 64.2 (CH), 37.2 (CH2), 31.2 (CH2); MS (CI) m/z (%) 215 [(MH+) − H2O]; HRMS calcd for C10H15O5: MH+, 215.0916. Found: MH+, 215.0919.Methyl (1S,3S,4S,6S,7R,9R)-9-hydroxy-7-(2′,3′-dihydroxy)propyloxy-3,4-dimethoxy-3,4-dimethyl-2,5-dioxabicyclo[4.4.0]decane-9-carboxylate (29)
To a stirred solution of allyl ether 26 (629 mg, 1.75 mmol) and N-methylmorpholine oxide (246 mg, 2.10 mmol) in 50% aqueous dioxane (20 ml) was added a freshly made aqueous solution of sodium tetraoxide (2.2 ml, 0.12 M). After stirring for 1 hour, ethyl acetate was added followed by saturated sodium bisulfite. The reaction mixture was stirred for 20 min. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (3 × 20 ml). The combined organic layers were dried (anhydrous Na2SO4) and concentrated in vacuo. The crude residue was purified by flash chromatography eluting with 10% methanol–ethyl acetate, and crystallised from ethyl ether–hexane to afford a 1.2 : 1 mixture of diastereoisomers of the glycerol29 (500 mg, 76%) as white needles. υmax (Nujol)/cm−1 3313b (OH) and 1735s (CO); δH (250 MHz, CDCl3): 4.33–4.22 (1 H, m), 3.89–3.53 (5 H, m), 3.73 (3 H, s), 3.24 (3 H, s), 3.23 (3 H, s), 2.22–1.83 (4H, m), 1.30 (3 H, s), 1.25 (3 H, s); δC (63 MHz, DEPT, CDCl3): 174.1 (C), 173.9 (C), 99.9 (C), 99.8 (C), 77.4 (CH), 77.3 (CH), 75.4 (C), 75.3 (C), 74.0 (CH2), 73.4 (CH), 73.0 (CH), 72.8 (CH2), 70.7 (CH), 69.9 (CH), 52.6 (OCH3), 47.9 (OCH3), 47.9 (OCH3), 38.8 (CH2), 38.7 (CH2), 36.5 (CH2), 36.4 (CH2), 17.5 (CH3), 17.4 (CH3); MS (CI) m/z (%) 363 [(MH+) − HOCH3]; HRMS calcd for C16H27O9: MH+, 363.1655. Found: MH+, 363.1650.(1S,3S,4R,5R)-1,4,5-Trihydroxy-3-(2′,3′-dihydroxy)propyloxycyclohexane-1-carboxylic acid (12)
A solution of acetal 29 (95 mg, 0.24 mmol) in a solution of trifluoroacetic acid–water (2 ml, 20 : 1 (v/v)) was stirred for 30 min and then the solvent was removed in vacuo. The crude residue was dissolved in an aqueous lithium hydroxide (3 ml, 0.5 M) and stirred for 1 hour. The aqueous layer was washed with diethyl ether (3 × 5 ml) and then was treated with Amberlite IR-120 until the pH was 6.0. The resin was filtered and washed with water. The filtrate was freeze-dried to afford a 1.2 : 1 mixture of diastereoisomers of the acid12 (60 mg, 94%) as an amorphous solid. υmax (Nujol)/cm−1 3430b (OH) and 1725s (CO); δH (300 MHz, D2O): 4.64 (1 H, dt, J = 10.1 and 4.5 Hz), 4.53 (1 H, q, J = 3.4 Hz), 4.49–4.41 (1 H, m), 4.29–3.97 (5 H, m), 2.49–2.03 (4 H, m); δC (75 MHz, DEPT, D2O): 181.3 (C), 81.6 (CH), 81.3 (CH), 78.4 (C), 77.3 (CH), 73.9 (CH2), 73.7 (CH2), 73.5 (CH), 73.4 (CH), 69.5 (CH), 65.5 (CH2), 43.3 (CH2), 43.2 (CH2), 36.7 (CH2), 36.5 (CH2); MS (CI) m/z (%) 249 [(MH+) − H2O]; HRMS calcd for C10H17O7: MH+, 249.0974. Found: MH+, 249.0967.Acknowledgements
Financial support from the Xunta de Galicia under project PGIDIT02RAG20901PR and Fundação para a Ciência e a Tecnologia is gratefully acknowledged.References
- C. Kleanthous, R. Deka, K. Davis, S. M. Kelly, A. Cooper, S. E. Harding, N. C. Price, A. R. Hawkins and J. R. Coggins, Biochem. J., 1992, 282, 687–695 CAS.
- C. Abell, in Enzymology and Molecular Biology of the Shikimate Pathway; Comprehensive Natural Products Chemistry, ed. U. Sankawa,Pergamon, Elsevier Science Ltd. Oxford, 1999, pp. 573–607 Search PubMed.
- H. C. Steinrucken and N. Amhrein, Biochem. Biophys. Res. Commun., 1980, 94, 1207–1212 CAS.
- N. H. Giles, M. E. Case, J. A. Baum, R. F. Geever, L. Huiet, V. B. Patel and B. M. Tyler, Microbiol. Rev., 1985, 49, 338–358 Search PubMed.
- (a) C. Kleanthous, M. Reilly, A. Cooper, S. Kelly, N. C. Price and J. R. Coggins, J. Biol. Chem., 1991, 266, 10893–10898 CAS; (b) R. K. Deka, C. Kleanthous and J. R. Coggins, J. Biol. Chem., 1992, 267, 22237–22242 CAS; (c) A. P. Leech, R. James, J. R. Coggins and C. Kleanthous, J. Biol. Chem., 1995, 270, 25827–25836 CrossRef CAS.
- (a) J. Harris, C. Kleanthous, J. R. Coggins, A. R. Hawkins and C. Abell, J. Chem. Soc., Chem. Commun., 1993, 1080–1081 RSC; (b) A. Shneier, J. Harris, C. Kleanthous, J. R. Coggins, A. R. Hawkins and C. Abell, Bioorg. Med. Chem. Lett., 1993, 3, 1399–1402 CrossRef CAS; (c) J. M. Harris, C. Gonzalez-Bello, C. Kleanthous, A. R. Hawkins, J. R. Coggins and C. Abell, Biochem. J., 1996, 319, 333–336 CAS.
- J. M. Harris, C. González-Bello, M. K. Manthey, J. R. Coggins and C. Abell, Bioorg. Med. Chem. Lett., 2000, 10, 407–409 CrossRef CAS.
- M. Frederickson, E. J. Parker, A. R. Hawkins, J. R. Coggins and C. Abell, J. Org. Chem., 1999, 64, 2612–2613 CrossRef CAS.
- D. G. Gourley, A. K. Shrive, I. Polikarpov, T. Krell, J. R. Coggins, A. R. Hawkins, N. W. Isaacs and L. Sawyer, Nat. Struct. Biol., 1999, 6, 521–525 CrossRef CAS.
- A. W. Roszak, D. A. Robinson, T. Krell, I. S. Hunter, M. Fredrickson, C. Abell, J. R. Coggins and A. Lapthorn, Structure, 2002, 10, 493–503 CrossRef.
- (a) S. B. Shuker, P. J. Hajduk, R. P. Meadows and S. W. Fesik, Science, 1996, 274, 1531–1534 CrossRef CAS; (b) P. J. Hajduk, G. Sheppard, D. G. Nettesheim, E. T. Olejniczak, S. B. Shuker, R. P. Meadows, D. H. Steinman, G. M. Carrera, P. A. Marcotte, J. Severin, K. Walter, H. Smith, E. Gubbins, R. Simmer, T. F. Holzman, D. W. Morgan, S. K. Davidsen and S. W. Fesik, J. Am. Chem. Soc., 1997, 119, 5818–5827 CrossRef CAS.
- (a) G. Jones, P. Willet and R. C. Glen, J. Mol. Biol., 1995, 245, 43–53 CrossRef CAS; (b) G. Jones, P. Willet, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS.
- T. Widlanski, S. L. Bender and J. R. Knowles, Biochemistry, 1989, 28, 7572–7582 CrossRef CAS.
- V. Van Rheenen, R. C. Kelly and D. Y. Cha, Tetrahedron Lett., 1976, 23, 1973–1976 CrossRef.
- G. Poli, Tetrahedron Lett., 1989, 30, 7385–7388 CrossRef CAS.
- J.-L. Montchamp, F. Tian, M. Hart and J. W. Frost, J. Org. Chem., 1996, 61, 3897–3899 CrossRef CAS.
- F. Tian, J.-L. Montchamp and J. W. Frost, J. Org. Chem., 1996, 61, 7373–7381 CrossRef CAS.
- R. Lakhamiri, P. Lhoste and D. Sinou, Tetrahedron Lett., 1989, 30, 4669–4672 CrossRef CAS.
- M. Dixon, Biochem. J., 1953, 55, 170–171 CAS.
- SYBYL. 6.5. 1699 South Hanley Road, St. Louis, Missouri, 63144, USA: Tripos Inc.
- WebLab ViewerPro4.0. 9685 Scranton Road, San Diego, CA 92121, USA: Accelrys Inc.
Footnote |
| † Electronic supplementary information (ESI) available: Dixon plots and results of docking experiments. See http://www.rsc.org/suppdata/ob/b3/b301731a/ |
| This journal is © The Royal Society of Chemistry 2003 |