
Towards promising oxoanion extractants: azacages and open-chain counterparts†
Abstract
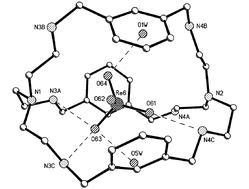
The efficiency of a series of amino-azacryptands for encapsulation and
- This article is part of the themed collection: Celebrating the 150th anniversary of the German Chemical Society
 Please wait while we load your content...
Please wait while we load your content...

