Changes in tropospheric composition and air quality due to stratospheric ozone depletion†
Keith R.
Solomon
a,
Xiaoyan
Tang
b,
Stephen R.
Wilson
c,
Prodromos
Zanis
d and
Alkiviadis F.
Bais
d
aCentre for Toxicology, University of Guelph, Guelph, ON, N1G 2W1, Canada
bPeking University, Center of Environmental Sciences, Beijing 100871, China
cDepartment of Chemistry, University of Wollongong, NSW, 2522, Australia
dAristotle University of Thessaloniki, Laboratory of Atmospheric Physics, Campus Box 149, Victoria, 54006 Thessaloniki, Greece
First published on 10th January 2003
Abstract
Increased UV-B through stratospheric ozone depletion leads to an increased chemical activity in the lower atmosphere (the troposphere). The effect of stratospheric ozone depletion on tropospheric ozone is small (though significant) compared to the ozone generated anthropogenically in areas already experiencing air pollution. Modeling and experimental studies suggest that the impacts of stratospheric ozone depletion on tropospheric ozone are different at different altitudes and for different chemical regimes. As a result the increase in ozone due to stratospheric ozone depletion may be greater in polluted regions. Attributable effects on concentrations are expected only in regions where local emissions make minor contributions. The vertical distribution of NOX (NO + NO2), the emission of volatile organic compounds and the abundance of water vapor, are important influencing factors. The long-term nature of stratospheric ozone depletion means that even a small increase in tropospheric ozone concentration can have a significant impact on human health and the environment.
Trifluoroacetic acid (TFA) and chlorodifluoroacetic acid (CDFA) are produced by the atmospheric degradation of hydrochlorofluorocarbons (HCFCs) and hydrofluorocarbons (HFCs). TFA has been measured in rain, rivers, lakes, and oceans, the ultimate sink for these and related compounds. Significant anthropogenic sources of TFA other than degradation HCFCs and HFCs have been identified. Toxicity tests under field conditions indicate that the concentrations of TFA and CDFA currently produced by the atmospheric degradation of HFCs and HCFCs do not present a risk to human health and the environment.
The impact of the interaction between ozone depletion and future climate change is complex and a significant area of current research. For air quality and tropospheric composition, a range of physical parameters such as temperature, cloudiness and atmospheric transport will modify the impact of UV-B. Changes in the chemical composition of the atmosphere including aerosols will also have an impact. For example, tropospheric OH is the ‘cleaning’ agent of the troposphere. While increased UV-B increases the OH concentration, increases in concentration of gases like methane, carbon monoxide and volatile organic compounds will act as sinks for OH in troposphere and hence change air quality and chemical composition in the troposphere. Also, changes in the aerosol content of the atmosphere resulting from global climate change may affect ozone photolysis rate coefficients and hence reduce or increase tropospheric ozone concentrations.
Introduction
Changes in air pollutants such as ozone (O3), nitrogen oxides (NOX), hydrogen peroxide (H2O2), formaldehyde (HCHO) and nitric acid (HNO3) in the lower atmosphere (troposphere) have important implications for human and environmental health. Understanding the factors that influence the concentrations of these pollutants is important for the protection of humans and their environment. The relationship between ultraviolet-B radiation (UV-B, 280–315 nm) and the photolysis of atmospheric trace gases such as O3, NO2, H2O2, HCHO, and HNO3 in the troposphere was reviewed in previous reports.1,2The previous report1,3 concluded that stratospheric ozone depletion causes changes in the chemical composition of the atmosphere through increased penetration of UV-B to the troposphere. Tropospheric ozone levels are sensitive to local concentrations of NOX and hydrocarbons. Increased UV-B is expected to increase the concentration of hydroxyl and other peroxy radicals and result in faster removal of pollutants. The effects of UV-B increases on tropospheric constituents like ozone, while not negligible, will be difficult to detect because the concentrations of these species are also influenced by many other variable factors, such as anthropogenic emissions.
The replacement of the ozone-depleting chlorofluorocarbon (CFC) refrigerants with less persistent products such as the hydrochlorofluorocarbons (HCFCs) and hydrofluorocarbons (HFCs) has also raised the question of the environmental significance of their breakdown products. In the previous report the environmental and human health significance of these breakdown products were not completely understood.1
Here we present a brief review of relevant new information. In addition, a discussion of interactions between tropospheric composition, air quality, changes due to ozone depletion, and climate change has been added. The conclusions are based on the current state of the science.
Changes in photodissociation rate coefficients due to stratospheric ozone depletion
Reductions in stratospheric ozone cause increased penetration of UV-B radiation to the troposphere and increases the photodissociation rates of tropospheric ozone and other trace gases such as NOX. The trends in photodissociation rates of tropospheric ozone and other chemical species were estimated by De Winter-Sorkina4,5 for the period 11/1978 to 4/1993, based on the TOMS/Nimbus-7 (version 7) total ozone data and Stratospheric Aerosol and Gas Experiment (SAGE) ozone trends together with experimental ozonesonde data. A three-dimensional chemical transport model was used to investigate the impact of changing photodissociation rates on tropospheric composition. The results showed that the trends in the daytime average photodissociation rate coefficient of ozone were + 18.0 ± 4.7% (95% confidence interval) per decade in February at the surface for zonal averages between 40 and 50°N. More importantly, there were also significant tropospheric ozone photodissociation trends of between +3 and 15% per decade at the surface at northern mid-latitudes in spring, summer and the first half of autumn. Significant long-term trends in tropospheric ozone photodissociation rate coefficients below 50°S were predicted throughout the year, peaking at +42 ± 15% per decade at the surface in October between 60 and 70°S. In general, the trends in ozone photodissociation rate coefficients due to stratospheric ozone depletion increased with altitude, reaching their maximum in winter-spring at about 8–12 km, the exact altitude depending on latitude. Overall, the trend in daytime average ozone photolysis rate coefficients is larger than the change in stratospheric ozone that caused them.De Winter-Sorkina5 found that the magnitude of the trends for the photodissociation rate coefficients for other atmospheric trace gases varied, although the regions and times of maximum impact were broadly the same as that for ozone photolysis. For example, at 40°N, the trends at the surface in January–April were calculated to be around +8% per decade for the photolysis of CH3CHO to produce CH3, + 6% per decade for HNO3 and + 2% per decade for H2O2, CH3OOH and N2O5. Once again, much larger changes are observed at 60 to 70°S with a trend of near 25% per decade for the photolysis reaction for CH3CHO listed above. The relative uncertainty in these estimates is similar to that of the ozone photolysis estimates.
Long-term global changes in tropospheric concentrations of ozone due to stratospheric ozone depletion have been calculated using the MOGUNTIA model5 by assuming that trace gas emissions remained constant. This gave a trend of + 2.4 ± 1.3% per decade for OH, −1.8 ± 1.3% per decade for CO and −0.8 ± 0.7% per decade for O3 and CH4. Thus, a small decrease in global tropospheric ozone was estimated due to stratospheric ozone reduction, with the tropospheric trends small or comparable to other factors, especially the changes due to anthropogenic emissions. The response of global OH concentrations to stratospheric ozone loss was found to be equivalent to the effect of a possible 10% NOX emission increase and 85% of this response was found to be equal to an estimated 6.5% CO decrease or a possible 10% tropical H2O increase.6
For all these chemical compounds, the observable impact of stratospheric ozone depletion on tropospheric trends can be overshadowed by local or regional emissions. Only in regions where local emissions make minor contributions will long-term effects due to stratospheric ozone reduction be clearly discernable from the changes due to other sources. However, the cumulative effect of long-term increases in ground level ozone concentrations may cause significant adverse environmental effects.
Changes in chemical composition of the troposphere due to stratospheric ozone depletion
The influence of increased UV-B photolysis on tropospheric chemical composition has been recognized more widely and has been examined in more detail recently through modeling and experimental studies. They include perturbations due to stratospheric ozone depletion as well as the influence of various tropospheric factors. The physical and chemical reactions involved in these processes have been reviewed previously.1 The net ozone production or loss depends mainly on the NOX concentration. Modeling studies suggest that an increase in UV-B (due to stratospheric ozone depletion) causes a decrease in tropospheric ozone in the clean environment of the southern hemisphere (very low NOX), and an increase in tropospheric ozone in the polluted areas of the northern hemisphere.7,8 The near UV photolysis of other chemical species such as HCHO (λ<330 nm) or CH3CHO (λ<330 nm), which are secondary sources of radicals, can also affect the surface ozone concentrations in an indirect way.9However, in certain cases, the photochemical link between total and surface ozone can be perturbed by weather-related atmospheric transport. For example, low total-column ozone could be related to upper troposphere/lower stratosphere anticyclones associated on the one hand with large scale subsidence of ozone rich air masses, and on the other hand with fair weather conditions with large ozone formation.10 Also, high total ozone column could be related to cut-off lows or upper troughs, synoptic systems associated with tropopause folds which may enhance the tropospheric ozone levels significantly within a few hours.11
Although the effects of change in UV intensity resulting from stratospheric ozone depletion on tropospheric chemistry are theoretically well understood from model calculations, there is a paucity of observational evidence. Stratospheric ozone is controlled by dynamical and chemical factors acting on different time scales and so the influence on surface UV-B and related tropospheric ozone chemistry should also act on different time scales. Long-term decreases in surface ozone in polar regions have been reviewed.1 The first observational evidence of changes in tropical tropospheric ozone associated with stratospheric ozone changes on a time scale of an 11 year solar cycle has been identified using the TCO (tropospheric column ozone) data derived from the TOMS satellite data.12
On a day-to-day basis, i.e., when circulation in the upper troposphere/lower stratosphere causes variations in total ozone column,13 there is even less observational evidence of the photochemical link between total and surface ozone due to the long lifetime of ozone and the fact that the photochemical link between total-column and surface ozone can be perturbed by dynamic coupling as discussed above. Thus, the photochemical influence of changing UV-B on in-situ ozone production and loss rate of 10% can easily be masked by transport effects, especially in late winter and at high latitudes.14 In addition, the sensitivity of a regional scale photochemistry model to changes in UV radiation, caused by moderate variations in either the total ozone column (25 DU) or in the aerosol optical depth, was found to be small (<0.5 nmol mol−1 for monthly average, June 1996).15 Only under sufficiently large changes of UV-B (that is, a large change in total ozone column) and suitable meteorological conditions can such an influence be detected, as presented in a case study on Swiss mountains in late winter14 and in a case study on Crete, Greece, during the PAUR II (Photochemical Activity and solar Ultraviolet Radiation) campaign.16
A modeling study by Ma and van Weele17 emphasized that influences on tropospheric pollutants are not isolated from interactions with other factors. It was found that the response of net ozone production to stratospheric ozone depletion was different at different altitudes and for different chemical coherent regimes. In addition to the surface concentration of NOX, the vertical distribution of NOX as well as the emissions of non-methane hydrocarbons (NMHCs), the abundance of water vapor, and ozone itself, are important factors. The threshold NOX concentration, at which the response to stratospheric ozone depletion of the net ozone production changes from negative to positive, depends on the chemical regime and varies during a day, but it is typically about 1 nmol mol−1 in a 24 h period. That is, it is around 100 times larger than the threshold NOX concentration for ozone production. Water vapour and NMHCs can further modulate tropospheric ozone production.
The importance of the vertical distribution of NOX on the net ozone production due to stratospheric ozone depletion in the polluted boundary region is illustrated in the observations of Brönnimann and co-authors14,18,19 at two elevated sites in Europe (Chaumont; 1,140 m above sea level (ASL), and Rigi;1,030 m ASL) where enhanced near-surface ozone concentrations were recorded. Mean diurnal cycles of ozone concentration showed a strong increase from late morning to late afternoon and, at the same time a decrease at the high alpine site Jungfraujoch (3,580 m ASL). The different diurnal ozone cycles can both be explained photochemically by taking into account the large difference in NOX concentrations (about two orders of magnitude). Brönnimann et al.18 also attempted to quantify the effect of changing UV-B radiation on surface ozone peaks in a day-to-day scale using a time series of measurement at the mountain sites. Seven years of ozone, NO and NOX data, meteorological measurements from Chaumont, total ozone and UV-B measurements from Arosa (1,847 m ASL), and surface albedo from satellite observations were investigated. The study was restricted to fair weather days with moderately high NOX concentrations. The estimated net effect on ozone peaks is normally within a range of 4 nmol mol−1, a range of about 6 nmol mol−1 is predicted for large UV-B changes. This is greater than 5% of the Swiss Air Quality 1 hour standard. Assuming that all other factors are constant, the trend due to the stratospheric ozone depletion observed in the last decades was found to be less than 0.12 nmol mol−1 yr−1. A 3-D mesoscale photochemical model (Metphomod) gave similar results to these measurements.20
The Metphomod model calculations were performed for the entire Swiss Plateau,20 and for the large stratospheric ozone depletion used in their calculation the changes in near ground level ozone was 0–3 nmol mol−1 (see Fig. 1). However, for the city of Zurich, they estimated changes of around +5 nmol mol−1 for both the winter and summer ‘ozone event’ they studied. In the urban airshed, such an increase could not simply be assigned, based on measurements, to stratospheric ozone depletion. However, it does represent a significant contribution to the overall ozone loading.
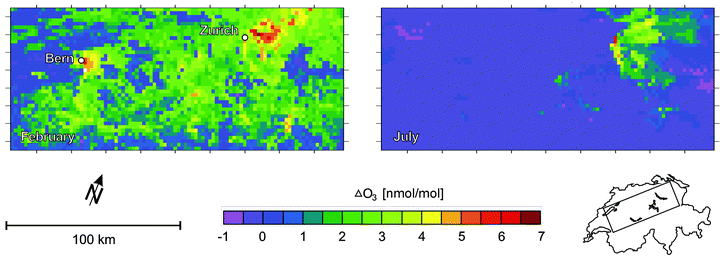 | ||
| Fig. 1 Ground level ozone production due to stratospheric ozone depletion. The calculations are for the region of Switzerland shown in the map. The left hand panel shows the calculated impact of ozone depletion from 400 DU to 240 DU for a simulation of an event in February 1998. The right hand panel shows a calculation for 30 July 1993, with an ozone decrease from 360 DU to 280 DU. (Figure based on the work of Brönnimann et al.20) | ||
Pirjola21 used a Lagrangian model to assess the interaction of elevated UV-B, volatile organic compound (VOC) emissions from plants (isoprene and some terpenes) and SO2. In the base case for emission rates of these organics, Pirjola reported a linear relationship between sulfate particle numbers and UV-B, with a 2.5-fold increase in particles for a 50% increase in UV-B. Under these conditions, the OH radical concentration increased by 9.8%. The concentration of biogenic organics is expected to increase with increasing CO2, and Pirjola has investigated this also. The model shows a decrease in OH and particle formation and an increase in ozone with an increase in organics. The increase in particle formation through a UV-B (XUVB(%)) enhancement can be offset by an increase in biogenic organics (YOrg(%)) as described by
| YOrg = 1.23 XUVB + 3.34 | (1) |
Humidity, temperature, SO2 emissions, and pre-existing particle concentrations were found to have little impact on the relationship.
The expected impact of stratospheric ozone depletion on formaldehyde (HCHO) is unclear, as both its production and destruction will be enhanced by ozone depletion. However, recent measurements of formaldehyde from Neumayer station in Antarctica22 found roughly twice the HCHO concentration in the spring during the ozone hole period than observed at a corresponding time in autumn. This suggests that, at least under Antarctic conditions, the concentration is enhanced by stratospheric ozone depletion.
Over the past few years, significant advances have been made in understanding how aerosols affect actinic fluxes and therefore tropospheric chemistry. This advance benefited greatly from the UV monitoring networks that have been put in place to monitor stratospheric ozone depletion. Dickerson et al.23 have shown that scattering by sulfate aerosols actually increases O3 production in the planetary boundary layer (PBL) of the eastern USA. Absorbing aerosols will reduce photolysis rates and reduce PBL-O3 production in polluted areas, but may actually reduce PBL-O3 loss under low NOX conditions, i.e. in more pristine regions.
Atmospheric production, fate, and effects of trifluoroacetic acid and related substances
The hydrochlorofluorocarbons (HCFCs) and hydrofluorocarbons (HFCs) HCFC-123, HCFC-124 and HFC-134a degrade to give trifluoroacetic acid (TFA).1 Use of HFCs may increase as the products are used to replace other products such as SF6, resulting in increased production of TFA. Environmental TFA can also be produced during the breakdown of other organofluorine compounds released to the atmosphere by human activities, e.g., halothane and isoflurane anaesthetics. TFA is also produced through the breakdown of TFM (a lampricide), trifluoromethyl-containing agrochemicals such as trifluralin, and Teflon.24,25 TFA is widely used in the chemical industry in processes where it is either consumed or becomes part of a chemical waste stream. A natural source of TFA has also been proposed from the weathering of fluorites that have been found to contain CF4.26 However, TFA has not yet been detected from these materials.Residues of TFA have been observed in water and air samples from many geographical areas (USA, Canada, Australia, South Africa, Germany, Israel, Ireland, France, Switzerland, Finland, and China) and show that TFA is a ubiquitous contaminant of the hydrosphere. Concentrations have been reported up to 0.04 mg L−1.27 Another halogenated acetic acid with similar properties to TFA is chlorodifluoroacetic acid (CDFA), which is also the breakdown product of HCFCs, and is formed via degradative reactions occurring in the atmosphere.28 Measured concentrations of CDFA are smaller than TFA. A maximum of 0.0002 ng L−1 was reported in water samples taken in Canada.28 Both of these halogenated acetic acids (HAAs) are persistent in the environment.
Fate and effects of TFA and CDFA in the ecosystem
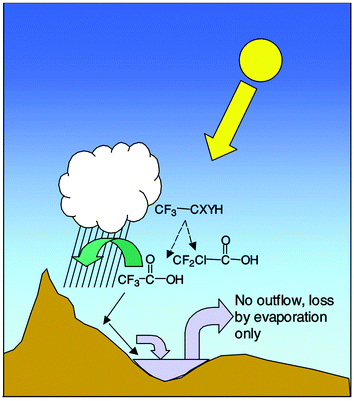 | ||
| Fig. 2 Illustration of the formation of persistent, water-soluble breakdown products of the HFCs and HCFCs (CF3–CXYH) and their movement and concentration by evaporation, along with other water-soluble salts to surface waters. | ||
Based on a no observed effect concentration (NOEC) of about 0.10 mg L−1 (as TFA) for the most sensitive standard algal species, Selenastrum capricornutum, a predicted no effect concentration in aquatic systems (PNECaqua) of 0.1 mg L−1 was proposed.1 Lack of effects of TFA on plants in aquatic microcosms under field conditions at concentrations up to 10 mg L−1 confirms low risk and the conservatism of the suggested PNECaqua.
On the basis of discussions in the previous review1 and more recent information, risks from TFA in the ecosystem are judged to be low. Although less data exist for CDFA, risks from this substance are also judged to be low.
Uncertainties as to the sources of TFA in the environment have been reduced through the identification of other pathways of formation. However, CDFA has been identified as a persistent degradation product of the HCFCs. CDFA is not considered toxic to aquatic plants but sensitivity of other species has not been reported. Although additional data on environmental concentrations of TFA (and CDFA) have been reported, continuing monitoring is recommended to confirm trends in surface and rainwater.
Interactions with global climate change
As discussed previously in this volume34 and more fully elsewhere,35,36 interactions between climate change and stratospheric ozone depletion may occur through many chemical, radiative and dynamic mechanisms. For example, changes in temperature will change reaction rates, variations in cloudiness will have a direct impact upon photolysis rates, and long-range transport can change the chemical composition. Several authors have reviewed potential interactions between ozone depletion and climate change.37–40 The review by Staehelin et al.38 concluded that the interactions between stratospheric ozone depletion and climate change would be a productive research domain in the future. Only a few examples of the possible interactions will be given here.Variations in ozone concentration, in particular in the upper troposphere and lower stratosphere, would be a crucial issue in the next decades because of links to climate change. Stratospheric ozone depletion leads to an increase in UV-B fluxes in the troposphere, which affect the photochemistry in the troposphere, especially the OH radicals. Increases in concentration of some greenhouse gases will act as sinks for OH in the troposphere, thus changing air quality and composition in the troposphere as suggested in the previous UNEP assessment1 and by Ma and van Weele.17 Studies by Prinn et al.41 using methylchloroform, suggest a recent decrease in global OH that may be due to anthropogenic emissions. An increase in atmospheric CO2 concentration would accelerate photosynthesis (CO2 fertilization) which might enhance the emissions of biogenic VOC's in forests and other natural ecological systems, and their oxidation reactions with hydroxyl radical which will act as a sink for OH.21
Other sources of tropospheric air pollutants may be affected by global warming. It is known that local and large-scale biomass fires such as are used for land-clearing are rich sources of NOX, CO, CH4, and other NMHCs, leading to enhanced tropospheric ozone production.42 During the 1994 South-East Asian haze episode (September–October 1994), measurements in Asia showed increased tropospheric concentrations of NOX (up to double) and ozone (∼20%).43 Besides extensive forest fires, local burning of biomass and fossil fuels also would have contributed to this. Thus, climate changes resulting from global warming may increase the risk of large-scale forest and brush-fires which affect concentrations of tropospheric air pollutants. Also, forest-fires induce haze aerosols that can increase multiple scattering, thus improving the efficiency of UV-B absorption of the boundary layer ozone. However, ozone production in the boundary layer may also be hindered by UV-B absorption by aerosols.44
Changes in the aerosol content of the atmosphere associated with global change may affect ozone photolysis rates45 and hence the tropospheric ozone levels. It was calculated that soot particles and mineral dust particles can reduce the ozone photolysis rate by 6% to 11%,45 and reductions of up to 50% have been estimated elsewhere.44,46 During the Sahara dust events in the PAUR II campaign, Balis et al.47 reported greatest effects on production of excited oxygen atoms (J(O1D)) during days with high aerosol content. Box model calculations for a case study during the same experiment at Crete were carried out. The model indicated that the presence of absorbing aerosols offset part of the increase in local net ozone production rate caused by the decrease in total column ozone concentration when enough NOX was present.16 A sensitivity analysis of photolysis rates and ozone production in the troposphere in response to aerosol properties used a coupled transport-chemistry-radiative transfer model,48 and reported reductions in ground level ozone concentrations of up to 70% in the presence of absorbing aerosols in the boundary layer (Fig. 3).
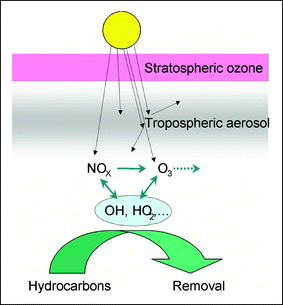 | ||
| Fig. 3 Illustration of the impact of tropospheric aerosols upon atmospheric chemistry. The aerosol can reduce the intensity of radiation, leading to a reduction in ozone production. Such a reduction offsets the impact of stratospheric ozone depletion. | ||
Although it is recognized that tropospheric ozone and other air pollutants are important with respect to human health and the environment, these pollutants are largely anthropogenic and concentrations vary in response to local human activity. The impact of the coupling of stratospheric ozone depletion and climate change on concentrations of pollutant will be difficult to quantify experimentally, even though it could be significant, especially in the regions where air quality has a large impact on human health.
Since global warming is expected to increase demand for refrigerants, the breakdown products of these substances are likely to increase in concentration in the troposphere at a greater than expected rate. While this could increase concentrations of persistent breakdown products such as TFA and CDFA, inputs to most environments would still be small enough (by a factor of about 1,000) that biologically significant concentrations would not be expected to occur in flowing surface waters. However, in areas where there is little or no outflow and increased evaporation due to increased surface temperatures, these products are expected to increase in concentration. While this may present a risk to aquatic organisms, these areas would also experience increases in concentrations of other water-soluble materials such as has already occurred in salt lakes and playas. The effects of these naturally occurring salts and other materials would likely be greater and more biologically significant than those of TFA or CDFA and would occur in the absence of these substances anyway. The results of this interaction with global climate change are judged to be of low significance.
References
- X. Tang, S. Madronich, T. Wallington and D. Calamari, Changes in tropospheric composition and air quality, J. Photochem. Photobiol. B., 1998, 46, 83–95 Search PubMed.
- D. L. Albritton, P. J. Aucamp, G. Megie and R. T. Watson, (Eds.), Scientific Assessment of Ozone Depletion: 1998, Global Ozone Research and Monitoring Project, World Meteorological Organization Report No. 44, Geneva.
- J. C. van der Leun, M. Tevini, X. Tang and R. C. Worrest, Environmental Effects of Ozone Depletion: 1998 UpdateUnited Nations Environment Programme, Nairobi, 1998, p. 192.
- R. De Winter-Sorkina, Impact of ozone layer depletion I: Ozone depletion climatology, Atmos. Environ., 2001, 35, 1609–1614 CrossRef CAS.
- R. De Winter-Sorkina, Impact of ozone layer depletion II: Changes in photodissociation rates and tropospheric composition, Atmos. Environ., 2001, 35, 1615–1625 CrossRef CAS.
- M. Krol, P. J. Van Leeuwen and J. Lelieveld, Global OH trend inferred from methylchloroform measurements, J. Geophys. Res., 1998, 103, 10697–10711 CrossRef CAS.
- A. M. Thompson, R. W. Stewart, M. A. Owens and J. A. Herwehe, Sensitivity of tropospheric oxidants to global chemical and climate change, Atmos. Environ., 1989, 23, 519–532 CrossRef CAS.
- S. Madronich and C. Granier, Impact of recent total changes on tropospheric ozone photodissociation, hydroxyl radicals, and methane trends, Geophys. Res. Lett., 1992, 19, 465–467 CAS.
- A. Ruggaber, R. Dlugi and T. Nakajimax, Modelling radiation quantities and photolysis frequencies in the troposphere, J. Atmos. Chem., 1994, 18, 171–210 CrossRef CAS.
- P. Zanis, E. Schuepbach, H. W. Gaeggeler, S. Huebener and L. Tobler, Factors controlling berrylium-7 at Jungfraujoch in Switzerland, Tellus, 1999, 51, 789–805 Search PubMed.
- G. Vaughan and G. D. Price, in Ozone in the atmosphere eds. R. D. Bojkov and P. Fabian, Deepak Publ, 1989, pp. 415–418 Search PubMed.
- S. Chandra, J. R. Ziemke and R. W. Stewart, An 11 year solar cycle in tropospheric ozone from TOMS measurements, Geophys. Res. Lett., 1999, 26, 185–188 CrossRef CAS.
- G. Vaughan and J. D. Price, On the relation between total ozone and meteorology, J. R. Meteorol. Soc., 1991, 117, 1281–1298 Search PubMed.
- S. Brönnimann and U. Neu, A possible photochemical link between stratospheric and near-surface ozone on Swiss mountain sites in late winter, J. Atmos. Chem., 1998, 31, 299–319 CrossRef CAS.
- J. E. Jonson, A. Kylling, T. Berntsen, I. S. A. Isaksen, C. S. Zerefos and K. Kourtidis, Chemical effects of UV fluctuations inferred from total ozone and tropospheric aerosol variations, J. Geophys. Res., 2000, 105, 14561–14574 CrossRef CAS.
- P. Zanis, K. Kourtidis, B. Rappenglueck, B. Balis, D. Melas, C. Zerefos, R. Schmitt, S. Rapsomanikis and P. Fabian, A case study on the link between surface ozone photochemistry and total ozone during the PAURII experiment at Crete—Comparison of observations with box model calculations, J. Geophys. Res., 2002, 107, 10.1029/2000JD000137 Search PubMed.
- J. Ma and M. van Weele, Effect of stratospheric ozone depletion on the net production of ozone in polluted rural areas, Chemosphere Global Change Sci., 2000, 2, 23–37 Search PubMed.
- S. Brönnimann, S. Voigt and H. Wanner, The influence of changing UVB radiation in near-surface ozone time series, J. Geophys. Res., 2000, 105, 8901–8913 CrossRef CAS.
- S. Brönnimann, Early spring ozone episodes: Occurrence and case study, Phys. Chem. Earth, 1999, 24, 531–536 Search PubMed.
- S. Brönnimann, W. Eugster and H. Wanner, Photo-oxidant chemistry in the polluted boundary layer under changing UV-B radiation, Atmos. Environ., 2001, 35, 3789–3797 CrossRef CAS.
- L. Pirjola, Effects of the increased UV radiation and biogenic VOC emissions on ultrafine sulfate aerosol formation, J. Aerosol Sci., 1999, 30, 355–367 CrossRef CAS.
- K. Riedel, R. Weller and O. Schrems, The effect of UV-radiation on photo-oxidants and formaldehyde in the atmosphere, in UV Radiation and its effects: An update, Eds. R. L. McKenzie, A. Reisinger and C. Watts, Royal Society of New Zealand, Christchurch, New Zealand, 2002 Search PubMed.
- R. Dickerson, S. Kondragunta, G. Stenchikov, K. Civerolo, B. Doddridge and B. Holben, The impact of aerosol on solar UV radiation and photochemical smog, Science, 1997, 278, 827–830 CrossRef CAS.
- D. A. Ellis, S. A. Mabury, J. W. Martin and D. C. G. Muir, Thermolysis of fluoropolymers as a potential source of halogenated organic acids in the environment, Nature, 2001, 412, 321–324 CrossRef CAS.
- D. Ellis and S. A. Mabury, The aqueous photolysis of TFM and related trifluoromethylphenols. An alternate source of trifluoroacetic acid in the environment, Environ. Sci. Technol., 2000, 34, 632–637 CrossRef CAS.
- J. Harnisch, M. Frische, R. Borchers, A. Eisenhauer and A. Jordan, Naturally fluorinated organics in fluorite and rocks, Geophys. Res. Lett., 2000, 27, 1883–1886 CrossRef CAS.
- D. Zehavi and J. N. Seiber, An analytical method for trifluoroacetic acid in water and air samples using headspace gas chromatographic determination of the methyl ester, Anal. Chem., 1996, 68, 3450–3459 CrossRef CAS.
- J. W. Martin, J. Franklin, M. L. Hanson, K. R. Solomon, S. A. Mabury, D. A. Ellis, B. F. Scott and D. C. G. Muir, Detection of chlorodifluoroacetic acid in precipitation: a possible product of fluorocarbon degradation, Environ. Sci. Technol., 2000, 34, 274–281 CrossRef CAS.
- B. R. Kim, M. T. Suidan, T. J. Wallington and X. Du, Biodegradability of trifluoroacetic acid, Environ. Eng. Sci., 2000, 17, 337–342 CAS.
- D. A. Ellis, M. L. Hanson, P. K. Sibley, T. Shahid, N. A. Fineburg, K. R. Solomon, D. C. G. Muir and S. A. Mabury, The fate and persistence of trifluoroacetic and chloroacetic acids in pond waters, Chemosphere, 2001, 42, 309–318 CrossRef CAS.
- M. L. Hanson, P. K. Sibley, K. R. Solomon, S. A. Mabury and D. C. G. Muir, Chlorodifluoroacetic acid (CDFA) fate and toxicity to the macrophytes Lemna gibba, Myriophyllum spicatum and Myriophyllum sibiricum in aquatic microcosms, Environ. Toxicol. Chem., 2001, 20, 2758–2767 CAS.
- M. Hanson, P. K. Sibley, D. Ellis, N. Fineberg, S. Mabury, K. R. Solomon and D. Muir, Trichloroacetic acid (TCA) fate and toxicity to the macrophytes Myriophyllum spicatum and Myriophyllum sibiricum under field conditions, Aquat. Toxicol., 2001, 56, 241–255 CrossRef.
- M. Hanson, P. K. Sibley, K. R. Solomon, S. Mabury and D. Muir, Trichloroacetic acid (TCA) and trifluoroacetic acid (TFA) mixture toxicity to the macrophytes Myriophyllum spicatum and Myriophyllum sibiricum in aquatic microcosms, Sci. Tot. Environ., 2002, 285, 247–259 Search PubMed.
- R. L. McKenzie, L. O. Bjorn, A. Bais and M. Ilyas, Changes in biologically active ultraviolet radiation reaching the Earth's surface, Photochemical and Photobiological Science, 2003, 2 Search PubMed , in press (DOI: 10.1039/b211155c).
- WMO (World Meteorological Organization), Scientific Assessment of Ozone Depletion: 2002, Global Ozone Research and Monitoring Project, World Meteorological Organization Report No. 47, Geneva, Switzerland, 1999.
- IPPC, Climate Change 2001: The Scientific Basis. Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, UK, 2001 Search PubMed.
- P. Taalas, J. Kaurola and A. Kylling, The impact of greenhouse gases and halogenated species on future solar UV radiation doses, Geophys. Res. Lett., 2000, 27, 1127–1130 CrossRef CAS.
- J. Staehelin, N. R. P. Harris and C. Appenzeller, Ozone trends: a review, Rev. Geophys., 2001, 39, 231–290 Search PubMed.
- D. T. Shindell, Climate and ozone response to increased stratospheric water vapor, Geophys. Res. Lett., 2001, 28, 1551–1554 Search PubMed.
- D. T. Shindell, G. A. Schmidt, R. L. Miller and D. Rind, Northern hemisphere winter climate response to greenhouse gas, ozone, solar and volcanic forcing, J. Geophys. Res., 2001, 106, 7193–7210 Search PubMed.
- R. G. Prinn, J. Huang, R. F. Weiss, D. M. Cunnold, P. J. Fraser, P. G. Simmonds, A. McCulloch, C. Harth, P. Salameh, S. O'Doherty, R. J. H. Wang, L. Porter and B. R. Miller, Evidence for substantial variations of atmospheric hydroxyl radicals in the past two decades, Science, 2001, 292, 1881–1888 CrossRef CAS.
- W. J. Collins, D. S. Stephenson, C. E. Johnson and R. G. Derwent, Trophospheric ozone in a global-scale three-dimensional langrangian mode and its response to NOx emission controls, J. Atmos. Chem., 1997, 26, 223–274 CrossRef CAS.
- M. Ilyas, A. Pandy and M. S. Jaafar, Changes to surface-level uvltaviolet-B radiation due to haze perturbation, J. Atmos. Chem., 2001, 40, 111–121 CrossRef CAS.
- J. P. Greenberg, A. B. Guenther, S. Madronich, W. Baugh, P. Ginoux, A. Druilhet, R. Delmas and C. Delon, Biogenic volatile organic compound emissions in central Africa during the Experiment for the Regional Sources and Sinks of Oxidants (EXPRESSO) biomass burning season, J. Geophys. Res., 1999, 104, 30659–30671 CrossRef CAS.
- H. Liao, Y. L. Yung and J. H. Seinfeld, Effects of aerosols on tropospheric photolysis rates in clear and cloudy atmospheres, J. Geophys. Res., 1999, 104, 23697–23707 CrossRef CAS.
- T. Castro, S. Madronich, S. Rivale, A. Muhlia and B. Mar, The influence of aerosols on photochemical smog in Mexico City, Atmos. Environ., 2001, 35, 1765–1772 CrossRef CAS.
- D. Balis, C. S. Zerefos, K. Kourtidis, A. F. Bais, A. Hofzumahaus, A. Kraus, R. Schmitt, K. Blumthaler and G. P. Gobbi, Measurements and modeling of the photolysis rates during the PAUR II campaign, J. Geophys. Res., 2002, 107 Search PubMed , in press.
- S. He and G. R. Carmichael, Sensitivity of photolysis rates and ozone production in the troposphere to aerosol properties, J. Geophys. Res., 1999, 104, 26307–26324 CrossRef CAS.
Footnote |
| † This article is published as part of the United Nations Environmental Programme: Environmental effects of ozone depletion and its interactions with climate change: 2002 assessment. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2003 |