Organically templated uranium(VI) sulfates: understanding phase stability using composition space
Paul M.
Thomas
,
Alexander J.
Norquist
,
Michael B.
Doran
and
Dermot
O'Hare
*
Inorganic Chemistry Laboratory, South Parks Road, University of Oxford, Oxford, UK OX1 3QR
First published on 12th November 2002
Abstract
Composition space has been used to study the phase stability of organically templated uranium sulfates in the UO2(CH3CO2)2·2H2O–1,3-diaminopropane–H2SO4 system. The syntheses and structures of two new compounds, [N2C3H12][UO2(H2O)(SO4)2] (USO-1) and [N2C3H12][(UO2)2(H2O)(SO4)3] (USO-2), are reported. The relative stability of the crystalline products in this system was found to be dependent upon the reactant mole fraction in the initial reaction gel, based upon the size and location of their respective crystallisation fields in the composition space.
Introduction
The reaction of inorganic species with organic templating agents under hydrothermal conditions is a versatile technique for the preparation of novel solid state materials.1 The structural diversity2 and desirable physical properties3 of these materials are well known. Despite the breadth of this chemistry, relatively few sulfate systems incorporating organic structure-directing agents have been reported.4 The hydrothermal chemistry of uranium-containing materials mirrors that of the transition metal and main group elements. A wide range of organically templated UIV and UVI materials has been prepared, incorporating fluoride,5 molybdate,6 phosphate,7 and phosphite,8 but the chemistry of organically templated uranium sulfates is unexplored.The investigation of this unknown chemistry will be performed in a systematic fashion, using composition space. Composition space9 and composition prisms10 directly relate the products to initial reactant mole fractions, by isolating these reaction variables. The concentrations of three reactants are varied systematically and a series of reactions are conducted using slightly different reactant mole fractions. All other variables, such as temperature, solvent concentration and reaction time are held constant. The results are plotted on a diagram to determine the areas of selective crystallisation and elucidate the factors governing product formation in hydrothermal crystallisations.
The syntheses and structures of two new uranium sulfates containing 1,3-diaminopropane (dap), designated USO-1 and USO-2 (uranium sulfate from Oxford), are reported. The UO2(CH3CO2)2·2H2O–dap–H2SO4 system has been explored and composition space used to relate the product composition to the initial reaction gel.
Experimental
Materials
1,3-Diaminopropane (99%, Aldrich) and sulfuric acid (98%, Aldrich) were used as received. Deionised water was also used in these syntheses. UO2Ac2·2H2O (Ac = CH3CO2−) was prepared6 from UO3 (99.8%, Strem).Methods
All reactions were conducted in poly[fluoro(ethylene/propylene)]-lined 23 mL autoclaves. The autoclaves were heated to 180 °C at 10 °C min−1, where the temperature was held constant for 24 h. Then, the reactions were cooled to room temperature at 6 °C h−1 and the autoclaves opened in air. The solid products were collected by filtration and washed with deionised water and acetone.Syntheses
X-Ray crystallographic analysis
Single crystals of dimensions 0.25 × 0.25 × 0.25 mm for USO-1 and 0.16 × 0.20 × 0.22 mm for USO-2 were used for the structural determination. Data were collected with an Enraf Nonius FR 590 Kappa CCD diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Crystals were mounted on a glass fibre using N-Paratone oil and cooled in situ to 150 K for data collection using an Oxford Cryostream 600 Series cooling system. Frames were collected, indexed and processed using Denzo SMN and the files scaled together using HKL GUI within Denzo SMN.11 The heavy atom positions were determined using SIR9712 and other non-hydrogen atoms refined using SHELXL-97.13 All non-hydrogen atom were located from Fourier difference maps and refined with anisotropic thermal parameters using full matrix least-squares procedures on Fo2 with I > 3σ(I). Hydrogen atoms attached to bound water molecules were located from Fourier difference maps, all others were placed in geometrically idealised positions. All calculations were performed using the programs CRYSTALS14 and CAMERON.15 Relevant crystallographic data are listed in Table 1 and selected atomic coordinates for USO-1 and USO-2 are given in Tables 2 and 3, respectively.| USO-1 | USO-2 | |
|---|---|---|
| a R 1 = Σ||Fo| − Fc||/Σ|Fo|. b wR 2 = [Σw(Fo2 − Fc2)2/[Σw(Fo2)2]1/2. | ||
| Formula | [N2C3H12][UO2(H2O)(SO4)2] | [N2C3H12][(UO2)2(H2O)(SO4)3] |
| Fw | 556.32 | 922.40 |
| Space group | P2/c (no. 13) | P21/n (no. 14) |
| a/Å | 7.2582(2) | 10.7391(3) |
| b/Å | 7.3697(2) | 10.3791(3) |
| c/Å | 11.8514(3) | 18.0265(7) |
| β/° | 99.4053(19) | 106.9424(12) |
| V/Å3 | 625.4 | 1922.1 |
| Z | 2 | 4 |
| D c/g cm−1 | 2.954 | 3.187 |
| μ/mm−1 | 13.368 | 17.238 |
| Reflections collected | 2404 | 7619 |
| Independent reflections | 1423 | 4301 |
| R 1 a | 0.0185 | 0.0343 |
| wR 2 b | 0.0434 | 0.0825 |
| Atom | x | y | z | U eq a |
|---|---|---|---|---|
| a U eq = (U11 + U22 + U33)/3. | ||||
| U(1) | 0.0 | 0.12121(2) | 0.25 | 0.0038 |
| S(1) | −0.12979(12) | −0.21813(11) | 0.02866(7) | 0.0070 |
| O(1) | −0.2454(4) | 0.1267(4) | 0.2147(2) | 0.0132 |
| O(2) | −0.0018(4) | −0.1375(4) | 0.1277(2) | 0.0117 |
| O(3) | −0.0172(4) | 0.2377(4) | 0.4353(2) | 0.0124 |
| O(4) | 0.0 | 0.4482(5) | 0.25 | 0.0181 |
| O(5) | −0.2917(4) | −0.1027(3) | −0.0076(2) | 0.0128 |
| O(6) | −0.1881(4) | −0.3980(3) | 0.0626(3) | 0.0140 |
| N(1) | −0.3718(4) | 0.2686(4) | −0.0436(3) | 0.0136 |
| C(1) | −0.4150(6 | 0.3774(5) | −0.1510(3) | 0.0150 |
| C(2) | −0.5 | 0.2565(8) | −0.25 | 0.0157 |
| Atom | x | y | z | U eq a |
|---|---|---|---|---|
| a U eq = (U11 + U22 + U33)/3. | ||||
| U(1) | 0.86449(3) | −0.25058(4) | 0.79153(2) | 0.0104 |
| U(2) | 1.37501(3) | −0.36209(3) | 0.76317(2) | 0.0084 |
| S(1) | 0.5945(2) | −0.4141(2) | 0.64626(16) | 0.0153 |
| S(2) | 0.9393(2) | −0.5652(2) | 0.72324(15) | 0.0127 |
| S(3) | 1.2011(2) | −0.1558(2) | 0.85600(14) | 0.0118 |
| O(1) | 0.8921(8) | −0.1838(9) | 0.7079(5) | 0.0246 |
| O(2) | 0.8382(8) | −0.3154(8) | 0.8755(5) | 0.0226 |
| O(3) | 1.0920(7) | −0.2449(7) | 0.8550(4) | 0.0174 |
| O(4) | 0.9094(7) | −0.0558(7) | 0.8635(4) | 0.0178 |
| O(5) | 0.6668(8) | −0.1389(9) | 0.7599(5) | 0.0291 |
| O(6) | 0.7046(8) | −0.3842(9) | 0.7157(6) | 0.0396 |
| O(7) | 0.9649(8) | −0.4489(8) | 0.7713(5) | 0.0295 |
| O(8) | 1.2263(7) | −0.3202(7) | 0.6954(4) | 0.0171 |
| O(9) | 1.5223(7) | −0.3996(7) | 0.8333(4) | 0.0164 |
| O(10) | 1.0609(7) | −0.6447(7) | 0.7495(4) | 0.0159 |
| O(11) | 0.4729(7) | −0.3713(7) | 0.6608(4) | 0.0185 |
| O(12) | 1.1636(7) | −0.0769(7) | 0.7849(4) | 0.0151 |
| O(13) | 1.2590(8) | −0.4921(8) | 0.8379(5) | 0.0261 |
| O(14) | 1.3165(6) | −0.2368(7) | 0.8571(4) | 0.0140 |
| O(15) | 0.609(1) | −0.3520(9) | 0.5783(6) | 0.0398 |
| O(16) | 0.9086(9) | −0.534(1) | 0.6418(5) | 0.0347 |
| O(17) | 1.2343(7) | −0.0773(7) | 0.9252(4) | 0.0200 |
| N(1) | 0.374(1) | 0.7839(11) | 0.0573(6) | 0.0304 |
| N(2) | 0.785(1) | 0.8563(11) | −0.0074(6) | 0.0281 |
| C(1) | 0.4924(12) | 0.7180(13) | 0.0460(8) | 0.0310 |
| C(2) | 0.5739(11) | 0.8169(11) | 0.0185(6) | 0.0193 |
| C(3) | 0.6982(12) | 0.7549(11) | 0.0131(7) | 0.0237 |
CCDC reference numbers 189594 and 189595.
See http://www.rsc.org/suppdata/jm/b2/b206694g/ for crystallographic data in CIF or other electronic format.
Infrared spectroscopy
All infrared measurements were obtained using a Perkin Elmer 1600 FT spectrometer. Samples were diluted with spectroscopic KBr and pressed into a pellet. Scans were run over the range 400–4000 cm−1.Thermogravimetric analysis
TGA measurements were conducted using a Rheometric Scientific STA 1500H thermal analyser. Samples were loaded into a platinum crucible and heated from ambient temperature to 800 °C at 10 °C min−1 under flowing argon.Elemental analysis
The Analytical Services Department of the Inorganic Chemistry Laboratory, University of Oxford conducted carbon, hydrogen, nitrogen, sulfur and uranium analyses.Results
USO-1
Two coordination polyhedra are observed in USO-1. The U6+ cation is seven-coordinate, in the form of a pentagonal bipyramid. The central [UO2]2+ U(1)–O(1) bond length, 1.762(2) Å, is close to the average for reported uranyl bonds in pentagonal bipyramidal coordination, 1.758(4) Å.16 The O(1)–U(1)–O(1) bond angle is 180°. The uranyl cation is bound to five oxide ligands, four of which are part of [SO4]2− tetrahedra. The average U–O bond length in the U–O–S linkages is 2.387 Å. A bound water molecule occupies the fifth equatorial coordination site. This U–O bond length is slightly longer, at 2.410(4) Å. Bond valence calculations17,18 on USO-1, using uranium parameters from Burns et al.,16 resulted in a value of 6.08 for U(1). Two of the four oxide ligands in the [SO4]2− tetrahedra bridge to the uranium, while the other two oxides are terminal. The bond lengths are 1.458(3), 1.468(3), 1.486(3) and 1.496(3) Å for the two terminal and two bridging oxides, respectively.One-dimensional [UO2(H2O)(SO4)2]2− chains are formed because each [SO4]2− tetrahedron links adjacent [UO7] pentagonal bipyramids (see Fig. 1). This chain topology is analogous to the mineral phases Mn[(UO2)(SO4)2(H2O)]·4H2O19 and [(UO2)(H2PO4)2(H2O)](H2O)2.20 Hydrogen bonding between the bound water and terminal oxides on the [SO4]2− tetrahedra link neighbouring chains, forming layers in the bc plane. The O(4)–O(6) hydrogen bond distance is 2.665(4) Å. The positively charged [dapH2]2+ templates reside between the layers, balancing the charge of the anionic chains. The structure is stabilised through extensive hydrogen bonding between the templates and one-dimensional chains (see Fig. 2).
![One-dimensional [UO2(H2O)(SO4)2]2− chains in USO-1. Tetrahedra and pentagonal bipyramids represent [SO4] and [UO7] moieties, respectively. Hydrogen bonds are shown as dashed lines.](/image/article/2003/JM/b206694g/b206694g-f1.gif) | ||
| Fig. 1 One-dimensional [UO2(H2O)(SO4)2]2− chains in USO-1. Tetrahedra and pentagonal bipyramids represent [SO4] and [UO7] moieties, respectively. Hydrogen bonds are shown as dashed lines. | ||
![Three-dimensional packing of USO-1. Tetrahedra and pentagonal bipyramids represent [SO4] and [UO7] moieties, respectively. Hydrogen atoms have been removed for clarity.](/image/article/2003/JM/b206694g/b206694g-f2.gif) | ||
| Fig. 2 Three-dimensional packing of USO-1. Tetrahedra and pentagonal bipyramids represent [SO4] and [UO7] moieties, respectively. Hydrogen atoms have been removed for clarity. | ||
The asymmetric stretch of the uranyl group was observed at 870 cm−1 in the IR spectrum. S–O and S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches were observed at 1000 and 1250 cm−1. The presence of the organic template was confirmed through the observation of characteristic N–H (3150 and 1650 cm−1) and C–H (2900 cm−1) bands.
O stretches were observed at 1000 and 1250 cm−1. The presence of the organic template was confirmed through the observation of characteristic N–H (3150 and 1650 cm−1) and C–H (2900 cm−1) bands.
A three-stage weight loss was observed in the thermogravimetric analysis of USO-1. The first corresponds to the loss of bound water molecules between 170–200 °C. The second weight loss represents the decomposition of the organic template between 380–450 °C (16.8%). The compound is completely calcined to UO2 by 500 °C (48.6%), as confirmed using powder X-ray diffraction.
USO-2
Two crystallographically distinct uranium sites are observed in USO-2. U(1) and U(2) are both seven-coordinate, consisting of two uranyl and five equatorial oxide ligands. The average uranyl bond length for U(1) and U(2) is 1.756 Å and the two O–U–O bond angles are 179.5(2) and 177.8(3)° for U(1) and U(2), respectively. The five equatorial coordination sites around U(1) are occupied by oxide ligands that are part of [SO4]2− tetrahedra. The average U(1)–Oeq bond length is 2.364 Å. The five equatorial coordination sites around U(2) are occupied by one bound water molecule and four sulfate oxides. The U(2)–OH2O bond distance is 2.484(7) Å and the average uranium to sulfate oxide bond distance is 2.377 Å. Bond valence calculations on USO-2, again using uranium parameters taken from Burns et al.,16 resulted in values of 6.27 and 6.09 for U(1) and U(2), respectively. Three crystallographically distinct sulfur sites are observed. Each [SO4]2− tetrahedron exhibits one terminal and three bridging oxide ligands.Two-dimensional [(UO2)2(H2O)(SO4)3]2− sheets are formed because each [SO4]2− tetrahedron links three adjacent [UO7] pentagonal bipyramids. The bound water on U(2) hydrogen bonds to an adjacent oxide ligand, the O(13)–O(10) distance is 2.764(8) Å (see Fig. 3). These anionic layers lie in the ab plane. [dapH2]2+ templates and occluded water reside between the layers and participate in extensive hydrogen bonding with both the layers and the water molecule bound to U(2) (see Fig. 4).
![Two-dimensional [(UO2)2(H2O)(SO4)3]2− layers in USO-2. Tetrahedra and pentagonal bipyramids represent [SO4] and [UO7] moieties, respectively. Hydrogen bonds are shown as dashed lines.](/image/article/2003/JM/b206694g/b206694g-f3.gif) | ||
| Fig. 3 Two-dimensional [(UO2)2(H2O)(SO4)3]2− layers in USO-2. Tetrahedra and pentagonal bipyramids represent [SO4] and [UO7] moieties, respectively. Hydrogen bonds are shown as dashed lines. | ||
![Three-dimensional packing of USO-2. Tetrahedra and pentagonal bipyramids represent [SO4] and [UO7] moieties, respectively. Selected hydrogen atoms have been removed for clarity.](/image/article/2003/JM/b206694g/b206694g-f4.gif) | ||
| Fig. 4 Three-dimensional packing of USO-2. Tetrahedra and pentagonal bipyramids represent [SO4] and [UO7] moieties, respectively. Selected hydrogen atoms have been removed for clarity. | ||
The characteristic uranyl group band was observed at 900 cm−1 in the IR spectrum with S–O and SO stretches at 970 and 1250 cm−1. Characteristic N–H (3250 cm−1) and C–H (2950 cm−1) bands were used to confirm the presence of the template, [dapH2]2+.
A three-stage weight loss was found in the thermogravimetric measurements. The initial weight loss of 3.5% between 230–280 °C is attributed to the loss of the bound water molecules in the lattice. Removal of the organic template between 320–390 °C results in a loss of 9.8%. Decomposition of the inorganic framework and removal of all sulfur-containing species was observed (24.7%) and is complete by 500 °C. Powder X-ray diffraction analysis of the calcined material was employed to confirm that decomposition led to crystalline UO2.
Composition space
The composition space diagram for the UO2Ac2·2H2O–dap–H2SO4 system (see Fig. 5) was constructed by conducting approximately 25 separate reactions, in which the initial reactant mole fractions were systematically varied. A constant amount (1 g) of solvent water was added to each reaction. Only part of the composition space has been explored because in other regions incomplete dissolution of starting materials and decomposition of the organic component results in poorly defined polycrystalline phases. The crystalline product from each reaction was isolated and analysed using powder X-ray diffraction to check the structure and determine phase purity. Four regions are observed. The first contains no solid reaction products, because all species remain soluble, and is located where the UO2Ac2·2H2O concentration is below 6% and the H2SO4 concentration is below 90%. If the resulting solution is left to slowly evaporate, solid products can be obtained, but they must be excluded from the composition space diagram because the both the reaction time and water concentration are different from the remainder of the space. Crystallisation in the second region is dominated by USO-1. This crystallisation field extends from approximately 60–84% H2SO4 and 20–28% dap mole fractions. USO-2 is the only crystalline phase present in the third region, which is found across the composition space diagram at dap mole fractions below 10%. The USO-1 and USO-2 crystallisation fields overlap in the fourth region, where the two phases co-crystallise. This region is approximately in the centre of the composition space diagram.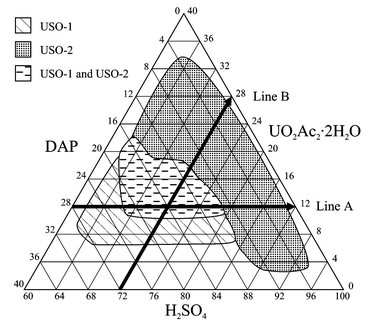 | ||
| Fig. 5 Composition space diagram for the UO2Ac2·2H2O–dap–H2SO4 system. The scale on each axis represents the relative mole fraction of the corresponding component. | ||
Discussion
The factors governing the composition of the crystalline products in the UO2Ac2·2H2O–dap–H2SO4 composition space diagram can be elucidated by comparing the series of reaction along the two lines shown in Fig. 5, denoted ‘Line A’ and ‘Line B.’Line A represents a series of reactions that were all conducted at the same UO2Ac2·2H2O mole fraction, 12%. USO-1 is the only crystalline phase observed at the dap-rich end of Line A. Mixtures of USO-1 and USO-2 are initially observed on moving toward the H2SO4-rich end of Line A. Finally, USO-2 becomes the only phase present. This can be explained by comparing the composition of USO-1 and USO-2. USO-1 has a U∶dap∶[SO4]2− ratio of 1∶1∶2, whereas the same ratio for USO-2 is 2∶1∶3. At the dap-rich end of Line A, USO-1 dominates the crystallisation because the dap∶[SO4]2− ratio more closely reflects the concentration ratio in the reaction gel. In regions where the dap concentration is high, the compound that incorporates more dap is more likely to be stable. The [SO4]2− concentration increases on moving along Line A toward the H2SO4 corner of the composition space diagram. The composition of the crystalline product reflects the change in the [SO4]2− concentration in solution. First, USO-2 is observed with USO-1, finally USO-2 is the only crystalline product. USO-2 is the stable product at high [SO4]2− concentrations just as USO-1 is stable at high dap concentrations.
Line B represents a series of reactions, all carried out at a constant H2SO4 mole fraction of 72%. No solid product is observed at the dap-rich end of Line B because the UO2Ac2·2H2O concentration is extremely low. First only USO-1 is observed, then it is present with USO-2 and, finally, USO-2 is the sole crystalline product at the UO2Ac2·2H2O-rich end of Line B. The shift in product composition can be explained by comparing the UO2Ac2·2H2O∶dap ratio in USO-1 (1∶1) and USO-2 (2∶1). USO-1 forms preferentially over USO-2 at the dap-rich end of Line B because its composition mirrors the conditions in solution, i.e. a low UO2Ac2·2H2O∶dap ratio. At the UO2Ac2·2H2O-rich end of Line B, only USO-2 forms because it has a higher UO2Ac2·2H2O∶dap ratio, which complements the composition of the reaction gel.
Conclusion
Two new organically templated uranium-containing compounds have been synthesised using H2SO4 and 1,3-diaminopropane. The [SO4]2− ion is incorporated into the new phases and also acts as a mineraliser. The factors governing the reaction product have been elucidated using composition space. The compositions of the crystalline phases obtained from hydrothermal reactions have been shown to be dependent upon the initial reactant mole fractions.Acknowledgement
The authors thank the EPSRC for support.References
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4, 756 CrossRef CAS; R. C. Haushalter and L. A. Mundi, Chem. Mater., 1992, 4, 31 CrossRef CAS; A. K. Cheetham, G. Férey and T. Loiseau, Angew. Chem., Int. Ed., 1999, 38, 3268 CrossRef CAS.
- T. E. Gier and G. D. Stucky, Nature, 1991, 349, 508 CrossRef CAS; G. Férey, J. Fluorine Chem., 1995, 72, 187 CrossRef CAS; M. I. Khan, L. M. Meyer, R. C. Haushalter, A. L. Schweitzer, J. Zubieta and J. L. Dye, Chem. Mater., 1996, 8, 43 CrossRef; S. Ayyappan, A. K. Cheetham, S. Natarajan and C. N. R. Rao, J. Solid State Chem., 1998, 139, 207 CrossRef CAS.
- D. W. Breck, Zeolite Molecular Sieves: Structure, Chemistry and Use, Wiley and Sons, London, 1974 Search PubMed; A. Clearfield, Chem. Rev., 1988, 88, 125 Search PubMed; P. B. Venuto, Microporous Mater., 1994, 2, 297 CrossRef CAS.
- M. I. Khan, S. Cervik and R. J. Doedens, Inorg. Chim. Acta, 1999, 292, 112 CrossRef CAS; A. Choudhury, J. Krishnamoorthy and C. N. R. Rao, Chem. Commun., 2001, 2610 RSC; I. Bull, P. S. Wheatley, P. Lightfoot, R. E. Morris, E. Sastre and P. A. Wright, Chem. Commun., 2002, 1180 RSC; G. Paul, A. Choudhury and C. N. R. Rao, Chem. Commun., 2002, 1904 RSC.
- R. J. Francis, P. S. Halasyamani and D. O'Hare, Angew. Chem., Int. Ed., 1998, 37, 2214 CrossRef CAS; R. J. Francis, P. S. Halasyamani, J. S. Bee and D. O'Hare, J. Am. Chem. Soc., 1999, 121, 1609 CrossRef; P. S. Halasyamani, S. M. Walker and D. O'Hare, J. Am. Chem. Soc., 1999, 121, 7415 CrossRef CAS; S. M. Walker, P. S. Halasyamani, S. Allen and D. O'Hare, J. Am. Chem. Soc., 1999, 121, 10513 CrossRef CAS; S. Allen, S. Barlow, P. S. Halasyamani, J. F. W. Mosselmans, D. O'Hare, S. M. Walker and R. I. Walton, Inorg. Chem., 2000, 39, 3791 CrossRef CAS; P. M. Almond, L. Deakin, M. J. Porter, A. Mar and T. E. Albrecht-Schmitt, Chem. Mater., 2000, 12, 3208 CrossRef CAS; C. E. Talley, A. C. Bean and T. E. Albrecht-Schmitt, Inorg. Chem., 2000, 39, 5174 CrossRef CAS; P. M. Almond, C. E. Talley, A. C. Bean, S. M. Peper and T. E. Albrecht-Schmitt, J. Solid State Chem., 2000, 154, 635 CrossRef CAS; P. M. Almond, L. Deakin, A. Mar and T. E. Albrecht-Schmitt, J. Solid State Chem., 2001, 158, 87 CrossRef CAS; P. M. Almond, L. Deakin, A. Mar and T. E. Albrecht-Schmitt, Inorg. Chem., 2001, 40, 886 CrossRef CAS; C. L. Cahill and P. C. Burns, Inorg. Chem., 2001, 40, 1347 CrossRef CAS.
- P. S. Halasyamani, R. J. Francis, S. M. Walker and D. O'Hare, Inorg. Chem., 1999, 38, 271 CrossRef CAS.
- R. J. Francis, M. J. Drewitt, P. S. Halasyamani, C. Ranganathachar, D. O'Hare, W. Clegg and S. J. Teat, Chem. Commun., 1998, 279 RSC; J. A. Danis, W. H. Runde, B. Scott, J. Fettinger and B. Eichhorn, Chem. Commun., 2001, 2378 RSC.
- M. Doran, S. M. Walker and D. O'Hare, Chem. Commun., 2001, 1988 RSC.
- P. Halasyamani, M. J. Willis, C. L. Stern, P. M. Lundquist, G. K. Wong and K. R. Poeppelmeier, Inorg. Chem., 1996, 35, 1367 CrossRef CAS; W. T. A. Harrison, L. L. Dussack and A. J. Jacobson, J. Solid State Chem., 1996, 125, 234 CrossRef CAS; A. J. Norquist, K. R. Heier, C. L. Stern and K. R. Poeppelmeier, Inorg. Chem., 1998, 37, 6495 CrossRef CAS.
- R. J. Francis, P. S. Halasyamani, J. S. Bee and D. O'Hare, J. Am. Chem. Soc., 1999, 121, 1609 CrossRef; S. M. Walker, P. S. Halasyamni, S. Allen and D. O'Hare, J. Am. Chem. Soc., 1999, 121, 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 513 CAS; A. J. Norquist, M. E. Welk, C. L. Stern and K. R. Poeppelmeier, Chem. Mater., 2000, 12, 1905 CrossRef CAS.
513 CAS; A. J. Norquist, M. E. Welk, C. L. Stern and K. R. Poeppelmeier, Chem. Mater., 2000, 12, 1905 CrossRef CAS. - Z. Otwinowski, Data Collection and Processing, Daresbury Laboratory, Warrington, UK, 1993 Search PubMed.
- G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997.
- D. J. Watkin, C. K. Prout, J. R. Carruthers, P. W. Betteridge and R. I. Cooper, CRYSTALS, Issue 11, Chemical Crystallography Laboratory, Oxford, UK, 2001.
- D. J. Watkin, C. K Prout and L. J. Pearce, CAMERON, Chemical Crystallography Laboratory, Oxford, UK, 1996.
- P. C. Burns, R. C. Ewing and F. C. Hawthorne, Can. Mineral., 1997, 35, 1551 Search PubMed.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect B., 1985, 41, 244 CrossRef.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect B., 1991, 47, 192 CrossRef.
- V. V. Tabachenko, V. L. Balashov, L. M. Kovba and V. N. Serezhkin, Koord. Khim., 1984, 10, 854 Search PubMed.
- E. Krogh-Anderson, I. G. Krogh-Anderson and G. Ploug-Soerensen, Solid State Protonic Conduct. Fuel Cells Sens., Eur. Workshop “Solid State Mater. Low Medium Temp. Fuel Cells Monit., Spec. Emphasis Proton Conduct.”, 3rd, 1985, 192 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |