
The first silyl- and germylboryl complexes: synthesis from novel (dichloro)silyl- and (dichloro)germylboranes, structure and reactivity
Abstract
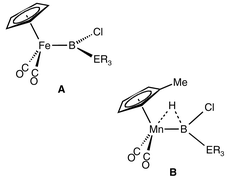
A series of silyl- and germyl(dichloro)boranes Cl2B–ER3 (5, ER3 = Si(SiMe3)3; 6, ER3 = Ge(SiMe3)3; 7, ER3 = Si(SiMe3)2–Si(SiMe3)3; 8, ER3 = SiPh3) was obtained in good yields from the corresponding bis(dimethylamino)boranes (Me2N)2B–ER3 (1, ER3 = Si(SiMe3)3; 2, ER3 = Ge(SiMe3)3; 3, ER3 = Si(SiMe3)2–Si(SiMe3)3; 4, ER3 = SiPh3) upon reaction with BCl3, and fully characterised in solution. These compounds proved to be versatile starting materials for the synthesis of the first silyl- and germylboryl complexes by salt elimination reactions starting from anionic transition metal complexes of the type Na[(η5-C5R5)Fe(CO)2] and K[(η5-C5R5)Mn(H)(CO)2]. The novel iron and manganese boryl complexes [(η5-C5R4R′)(OC)2Fe–B(Cl)Si(SiMe3)3] (9a, R = R′ = H; 9b, R = H, R′ = Me; 9c, R = R′ = Me), [(η5-C5H5)(OC)2Fe–B(Cl)Ge(SiMe3)3] (9d), [(η5-C5H5)(OC)2Fe–B(Cl)Si(SiMe3)2Si(SiMe3)3] (9e), [(η5-C5H4Me)(OC)2(H)Mn–B(Cl)Si(SiMe3)3] (10a) and [(η5-C5H4Me)(OC)2(H)Mn–B(Cl)Ge(SiMe3)3] (10b) were obtained in yields between 35 and 70%, fully characterised in solution and in the case of 9a, d, and 10a also in the crystalline state. They are all characterised by metal–boron σ-bonds without indication of any metal-to-boron π-backdonation. There is, however, spectroscopical and structural evidence for the presence of a Mn–H–B bridge in the case of the manganese complexes 10a, b. The first transhalogenation of a metal coordinated boryl ligand was achieved by reaction of 9a with TlF affording the fluoroboryl complex [(η5-C5H5)(OC)2Fe–B(F)Si(SiMe3)3] (11), which was characterised in solution and in the solid state.
- This article is part of the themed collection: Celebrating the 150th anniversary of the German Chemical Society
 Please wait while we load your content...
Please wait while we load your content...