Identification of Tyr58 as the proton donor in the aspartate-α-decarboxylase reaction
S. Adrian Saldanha†a, Louise M. Birch†a, Michael E. Webba, Brent K. Nabbsa, Frank von Delftb, Alison G. Smithc and Chris Abell*a
aUniversity Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: ca26@cam.ac.uk
bDepartment of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge, UK CB2 1GA
cDepartment of Plant Sciences, University of Cambridge, Downing Street, Cambridge, UK CB2 3EA
First published on 29th August 2001
Abstract
The decarboxylation of L-aspartate by E. coliL-aspartate-α-decarboxylase (ADC) is shown to occur with retention of configuration; analysis of the protein structure identifies Tyr58 as the proton donor in the decarboxylation mechanism.
ADC catalyses the decarboxylation of L-aspartate to β-alanine.1 It is one of four enzymes in E. coli involved in the biosynthesis of pantothenate (vitamin B5),2 the precursor of 4′-phosphopantetheine and coenzyme A. ADC is a tetrameric protein comprising four 14 kDa subunits. The X-ray crystal structure of E. coli ADC has been determined, and shows that the active sites are between subunits.3
The mechanism of decarboxylation involves formation of an imine between the amino group of aspartate and an integral pyruvoyl group. The pyruvoyl group is formed by an autocatalytic post-translational modification which cleaves the Gly24–Ser25 bond and converts Ser25 into the pyruvoyl group. This reaction is analogous to that which occurs in histidine decarboxylase4 and S-adenosylmethionine decarboxylase.5
The proposed enzyme mechanism is depicted in Scheme 1. After formation of the enzyme–substrate adduct (1), carbon dioxide is lost to form the extended enolate (2). This then reprotonates to form the imine adduct of the product β-alanine (3), which is finally released by hydrolysis to regenerate the pyruvoyl group. Evidence for this mechanism comes from trapping of the enzyme–substrate adduct and enzyme–product adduct by reduction with cyanoborohydride, and subsequent detection by electrospray mass spectrometry.6
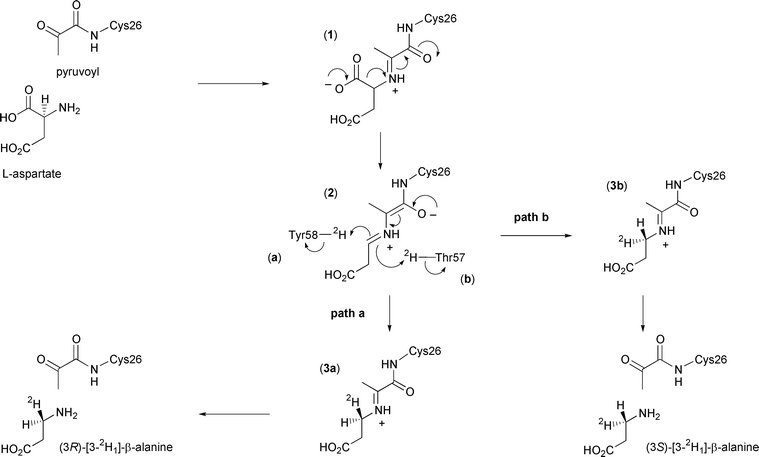 | ||
| Scheme 1 Catalytic mechanism of ADC showing the possible stereochemical outcomes in the presence of 2H2O. | ||
In this paper we report the results of our investigation into the stereochemical course of the decarboxylation reaction catalysed by ADC. Analysis of the crystal structure of ADC suggests that the proton may come from the phenolic group of Tyr58 or the hydroxy group of Thr57, which lie above and below the face of the enzyme–intermediate adduct (2) respectively (Fig. 1). If the proton donor were Tyr58, the proton would be added to the re face (Scheme 1, path a), corresponding to decarboxylation–protonation occurring with overall retention of configuration. Conversely, decarboxylation–protonation would occur with inversion of configuration if the proton were added from Thr57 to the si face of (2) (path b). Alternatively, but less probable, the reprotonation could be non-stereospecific.
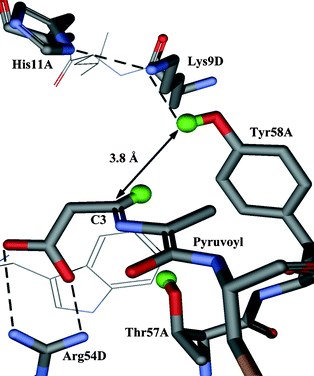 | ||
| Fig. 1 Active site of ADC with the enzyme-intermediate adduct (2) modelled in. The active site is at the interface of subunits and is composed of residues from both subunits (in this case subunits A and D). | ||
The stereochemical course of the reaction can be determined by using ADC to catalyse the conversion of L-aspartate into β-alanine in deuterium oxide. Stereospecific deuteriation of (2) will generate chirally-labelled β-alanine which can be characterised as its camphanoyl derivative by comparison with previously assigned spectra of (3R)- and (3S)-N-camphanoyl-[3-2H1]-β-alanine.7
The panD gene6,8 in E. coli was overexpressed with an N-terminal hexa-histidine tag to facilitate protein purification by affinity chromatography on a nickel–nitrilotriacetic acid (Ni–NTA) column. L-Aspartate (50 mg) and His6-ADC (6 mg) were incubated in phosphate buffer (pD = 7.6, 50 mM) in 2H2O for four days at 25 °C. The enzyme was removed by ultrafiltration and the solution lyophilised to dryness. The residue was derivatised with camphanoyl chloride by shaking in a biphasic mixture of 2 M NaOH and toluene for 3 h, followed by extraction with chloroform, drying with Na2SO4 and evaporation. Unlabelled N-camphanoyl-β-alanine was also synthesised by this procedure. The 1H NMR spectra of the C3 protons of N-camphanoyl-β-alanine and the camphanoyl derivative of the biotransformed material are shown in Fig. 2.9
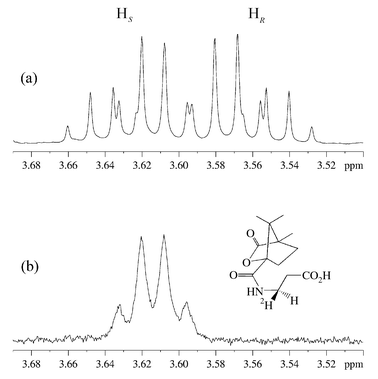 | ||
| Fig. 2 1H NMR (500 MHz, C2HCl3) of the C3 protons of β-alanine in (a) N-camphanoyl-β-alanine and (b) camphanoyl derivative of the biotransformed material. | ||
Comparison of the two spectra in Fig. 2 shows that a deuterium atom has been incorporated, leading to loss of the signal for one proton. The geminal 1H coupling to the remaining proton has been replaced by a smaller 2H coupling, causing an apparent simplification of the multiplet and broadening of the signals. This confirms that the reprotonation is stereospecific.
In order to determine the absolute stereochemistry of the labelling of β-alanine, the spectra were compared to published data on (3R)- and (3S)-N-camphanoyl-[3-2H1]-β-alanine, determined as part of a study on the dihydropyrimidine dehydrogenase reaction.7 The upfield multiplet in Fig. 2a has been assigned to the 3R proton. The biotransformed compound, in which this proton has been replaced by a deuteron, is consequently (3R)-N-camphanoyl-[3-2H1]-β-a lanine. This is the expected product if deuterium is delivered from Tyr58 to the re face of (2), with the decarboxylation–protonation steps proceeding with overall retention of configuration (Scheme 1, path a).
The enzyme–intermediate adduct (2) has been modelled into the crystal structure of ADC (Fig. 1). In this model the distance between the hydroxy proton of Thr57 and the C3 carbon is 3.3 Å. The corresponding distance from C3 to the phenolic proton of Tyr58 is 3.8 Å. Despite being the closest potential donor, Thr57 may not be the most appropriate. It is relatively buried in the active site, and deprotonation of the hydroxy group could destabilise the neighbouring salt bridge between the β-carboxyl of the enzyme–intermediate adduct (2) and Arg54. Tyr58 appears to be better placed; in particular it is part of a potential proton relay with Lys9 and His11. All three are conserved residues. As His11 is close to the surface of the protein it could acquire a proton from the solvent.
In conclusion, the stereochemical labelling study and analysis of a model of the putative intermediate bound to the enzyme (2), together suggest that the source of the proton in the reprotonation step is the phenolic group of Tyr58.
Acknowledgements
We would like to thank Drs Martyn Frederickson, Peter Grice and Nick Bampos and the NMR technical staff for their assistance.Notes and references
- J. M. Williamson and G. M. Brown, J. Biol. Chem., 1979, 254, 8074 CAS.
- G. M. Brown and J. M. Williamson, Adv. Enzymol. Relat. Areas Mol. Biol., 1982, 53, 345 Search PubMed.
- A. Albert, V. Dhanaraj, U. Genschel, G. Khan, M. K. Ramjee, R. Pulido, B. L. Sibanda, F. von Delft, M. Witty, T. L. Blundell, A. G. Smith and C. Abell, Nat. Struct. Biol., 1998, 5, 289 CrossRef CAS.
- T. Gallagher, E. E. Snell and M. L. Hackert, J. Biol. Chem., 1989, 264, 12737 CAS.
- G. D. Markham, C. W. Tabor and H. J. Tabor, J. Biol. Chem., 1982, 257, 2063.
- M. K. Ramjee, U. Genschel, C. Abell and A. G. Smith, Biochem. J., 1997, 323, 661 CAS.
- D. Gani and D. W. Young, J. Chem. Soc., Perkin Trans. 1, 1985, 7, 1355 RSC.
- J. E. Cronan, J. Bacteriol., 1980, 141, 1291 CAS.
- N-Camphanoyl-β-alanine: 1H NMR δ (500 MHz, C2HCl3, β-alanine signals) 3.63 (H, m, 3-HS), 3.56 (H, m, 3-HR), 2.61 (2H, t, J = 5.9 Hz, CH2); 13C NMR δ (125.8 MHz, C2HCl3) 178.4, 175.9, 167.4, 92.4, 55.2, 53.9, 34.6, 33.8, 30.1, 28.9, 16.6, 16.3, 9.6; HRMS (ES) calcd for C13H19NaNO5 (MNa) m/z 292.1161, found m/z 292.1169. (3R)-N-Camphanoyl-[3-2H1]-β-a lanine: 1H NMR δ (500 MHz, C2HCl3, β-alanine signals) 3.61 (H, m, 3-HS), 2.63 (2H, d, J = 5.9 Hz, CH2); 13C NMR δ (125.8 MHz, C2HCl3) 178.4, 176.2, 167.3, 92.4, 55.2, 53.9, 34.4, 33.9, 30.1, 29.0, 16.6, 16.4, 9.6; HRMS (ES) calcd for C13H18DNaNO5 (MNa) m/z 293.1224, found m/z 293.1212..
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2001 |