
Exploiting the capabilities of quantum chemical simulations to characterise the hydration of molecular compounds
Abstract
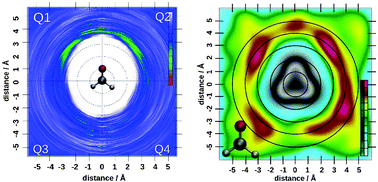
Solvation phenomena have been the focus of experimental and theoretical research for many decades. While the determination of solvation properties of mono-atomic, ionic species is already a challenging task, the associated difficulties are amplified in the case of molecular compounds. Theoretical approaches have the ability to yield manifold information of the investigated systems, providing that a reliable description of the interatomic energies and forces is employed. Hybrid quantum mechanical/molecular mechanical simulations constitute a particularly successful approach for the study of solvation phenomena, and have been employed in investigations of a variety of molecular compounds in aqueous solution. While an accurate description of these systems depends on the application of reliable, theoretical approaches, the analysis of the resulting dataset is a challenge of its own. It is outlined that standard means of analysis via pair correlation functions may yield an unspecific picture of hydration phenomena, and in some cases wrong conclusions on the hydration may be drawn. More advanced means of structural analysis are thus required as they yield manifold information on solvation phenomena since pair correlations are not sufficiently responsive in such cases. The theoretical approaches required to obtain a reliable description of the investigated systems as well as increasingly complex means of structural analysis ranging from two- and three-body correlations via angular-radial distributions and two-dimensional particle mapping to the so-called SLICE projections are discussed. The application of these analysis schemes is demonstrated for a variety of systems including mono- and polyatomic ionic solutes as well as molecular organic compounds, highlighting various aspects of the hydration of these species.
 Please wait while we load your content...
Please wait while we load your content...

