
Thiol-Michael addition in polar aprotic solvents: nucleophilic initiation or base catalysis?†
 a
Filip E.
Du Prez
c
and
a
Filip E.
Du Prez
c
and
Abstract
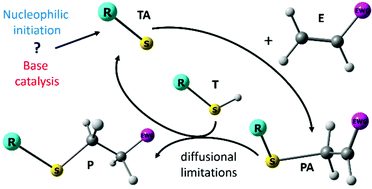
The thiol-Michael addition of ethanethiol to ethyl acrylate, methyl vinylsulfone and maleimide initiated by ethyl-, diethyl-, triethylamine and triethylphosphine in tetrahydrofuran (THF) is investigated at room temperature for concentrations ranging from 0.5 to 2 mol L−1 for the reactants and 0.03 to 0.3 mol L−1 for the initiators. Rate coefficients for all elementary steps in a reaction scheme consisting of both the base catalyzed and the nucleophile initiated mechanism are calculated using CBS-QB3 corrected for solvation with COSMO-RS. Diffusional limitations are taken into account using the coupled encounter pair model. The ab initio apparent kinetic parameters are used in a microkinetic model and simulated conversions agree well with experimental data. Competition with the aza-Michael addition is shown to be insignificant. Regardless of the choice of ene or catalyst, conversion is governed by an anionic cycle in which first an addition from the thiolate to the ene occurs, followed by a rate-controlling proton transfer to the obtained Michael adduct anion from another thiol. For acrylates and vinylsulfones, the addition of the thiolate to the ene is quasi-equilibrated, while for maleimides this elementary reaction has a positive affinity, explaining their large reactivity. The choice of catalyst or ene strongly affects the initiation mechanism. Using tertiary phosphines only nucleophilic initiation takes place while with tertiary amines, only base catalysis occurs. For primary and secondary amines both initiation mechanisms contribute. The presented kinetic parameters and the insights on diffusional limitations are critical for the further optimization of thiol-Michael additions for polymer conjugation.
 Please wait while we load your content...
Please wait while we load your content...

