
A structural model of the copper ATPase ATP7B to facilitate analysis of Wilson disease-causing mutations and studies of the transport mechanism†
Abstract
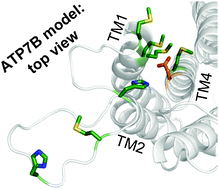
The copper-transporting ATPase ATP7B has an essential role in human physiology, particularly for the liver and brain function. Inactivation of ATP7B is associated with a severe hepato-neurologic disorder, Wilson disease (WD). Hundreds of WD related mutations have been identified in ATP7B to date. The low frequency and the compound-heterozygous nature of causative mutations complicate the analysis of individual mutants and the establishment of genotype–phenotype correlations. To facilitate studies of disease-causing mutations and mechanistic understanding of WD, we have homology-modelled the ATP7B core (residues 643–1377) using the recent structure of the bacterial copper-ATPase LCopA as a template. The model, supported by evolutionary conservation and hydrophobicity analysis, as well as existing and new mutagenesis data, allows molecular interpretations of experimentally characterized clinical mutations. We also illustrate that structure and conservation can be used to grade potential deleterious effects for many WD mutations, which were clinically detected but have not yet been experimentally characterized. Finally, we compare the structural features of ATP7B and LCopA and discuss specific features of the eukaryotic copper pump.
- This article is part of the themed collection: Metals and Genetics
 Please wait while we load your content...
Please wait while we load your content...