
Binding free energy based structural dynamics analysis of HIV-1 RT RNase H–inhibitor complexes†

Abstract
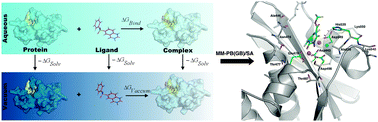
Accurate prediction of binding free energies associated with small molecules binding to a receptor is a major challenge in drug design processes. To achieve this goal many computational methods have been developed ranging from highly efficient empirical based docking schemes to high accuracy methods based on e.g. free energy calculations. In this study, binding affinity predictions for a set of HIV-1 RNase H inhibitors have been performed using MM-PB(GB)/SA methods. The current study describes in detail how the choice of initial ligand structures, e.g. protonation states, impacts the predicted ranking of the compounds. In addition we study the structural dynamics of the RNase H complexes using molecular dynamics. The role of each residue contribution to the overall binding free energy is also explored and used to explain the variations in the inhibition potency. The results reported here can be useful for design of small molecules against RNase H activity in the development of effective drugs for HIV treatment.
- This article is part of the themed collection: Computational Integrative biology (IB)
 Please wait while we load your content...
Please wait while we load your content...