
Spectroscopic and computational studies of reversible O2 binding by a cobalt complex of relevance to cysteine dioxygenase†

Abstract
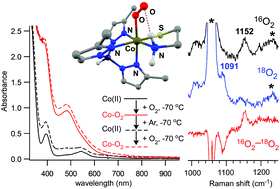
The substitution of non-native metal ions into metalloenzyme active sites is a common strategy for gaining insights into enzymatic structure and function. For some nonheme iron dioxygenases, replacement of the Fe(II) center with a redox-active, divalent transition metal (e.g., Mn, Co, Ni, Cu) gives rise to an enzyme with equal or greater activity than the wild-type enzyme. In this manuscript, we apply this metal-substitution approach to synthetic models of the enzyme cysteine dioxygenase (CDO). CDO is a nonheme iron dioxygenase that initiates the catabolism of L-cysteine by converting this amino acid to the corresponding sulfinic acid. Two mononuclear Co(II) complexes (3 and 4) have been prepared with the general formula [Co2+(TpR2)(CysOEt)] (R = Ph (3) or Me (4); TpR2 = hydrotris(pyrazol-1-yl)borate substituted with R-groups at the 3- and 5-positions, and CysOEt is the anion of L-cysteine ethyl ester). These Co(II) complexes mimic the active-site structure of substrate-bound CDO and are analogous to functional iron-based CDO models previously reported in the literature. Characterization with X-ray crystallography and/or 1H NMR spectroscopy revealed that 3 and 4 possess five-coordinate structures featuring facially-coordinating TpR2 and S,N-bidentate CysOEt ligands. The electronic properties of these high-spin (S = 3/2) complexes were interrogated with UV-visible absorption and X-band electron paramagnetic resonance (EPR) spectroscopies. The air-stable nature of complex 3 replicates the inactivity of cobalt-substituted CDO. In contrast, complex 4 reversibly binds O2 at reduced temperatures to yield an orange chromophore (4-O2). Spectroscopic (EPR, resonance Raman) and computational (density functional theory, DFT) analyses indicate that 4-O2 is a S = 1/2 species featuring a low-spin Co(III) center bound to an end-on (η1) superoxo ligand. DFT calculations were used to evaluate the energetics of key steps in the reaction mechanism. Collectively, these results have elucidated the role of electronic factors (e.g., spin-state, d-electron count, metal–ligand covalency) in facilitating O2 activation and S-dioxygenation in CDO and related models.
- This article is part of the themed collection: Frontiers in Spectroscopic Techniques in Inorganic Chemistry
 Please wait while we load your content...
Please wait while we load your content...

