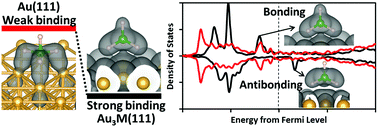
We study the adsorption of borohydride on Au and Au-based alloys (Au3M with M = Cr, Mn, Fe, Co, and Ni) using first-principles calculations based on spin-polarized density functional theory. Favorable molecular adsorption and greater adsorption stability compared to pure Au are achieved on Au3M alloys. For these alloys, there is an emergence of unoccupied states in the surface d band around the Fermi level with respect to the fully occupied d band of pure Au. Thus, the derived antibonding state of the sp–d interaction is upshifted and becomes unoccupied compared to pure Au. The B–H bond elongation of the adsorbed borohydride on these alloy surfaces points to the role of surface-parallel (dxy and dx2−y2 states) components of the d-band of the alloying metal M, most pronouncedly in the cases of M = Co or Ni. On the alloy surfaces, B binds directly with the alloying metal, unlike in the case of pure Au where the surface bonding is through the H atoms. These results pose relevant insights into the design of Au-based anode catalysts for the direct borohydride fuel cell.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


