
Intimate electronic coupling in cationic homodimeric iridium(iii) complexes†
Abstract
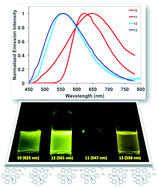
Herein, we report two new cationic iridium(III) homodinuclear structures linked through a diyne moiety at the 5-position of the bipyridyl ligand (1,4-di(2,2′-bipyridin-5-yl)buta-1,3-diyne) and compare these to mononuclear model systems bearing a 5-ethynyl-2,2′-bipyridine ligand. Low energy bands observed in the absorption spectra point to charge-transfer transitions for all four complexes, with these bands red-shifted in the case of the two dinuclear complexes. Electrochemical studies show metal-centred oxidation and ligand-centred first reduction potentials. In the case of the dimer bearing 2-phenylpyridine (ppyH) cyclometallating ligands, cyclic voltammetry (CV) measurements reveal two one-electron oxidation waves and a corresponding reduction in the HOMO–LUMO gap (ΔEred-ox) compared to a mononuclear system, pointing to a significant electronic coupling between the two iridium(III) metals. The room temperature emission spectrum of this dimer is also bathochromically shifted, corroborating the CV data. In the case of the iridium dimer bearing 2-(2,4-difluorophenyl)-5-methylpyridine (dFMeppy) ligands, only a single one-electron oxidation wave is observed, but with the expected smaller ΔEred-ox value, compared to its mononuclear counterpart. The emission spectra at room temperature are generally broad and featureless with only modest quantum efficiencies (ΦPL = 1.4–8.4%) in 2-methyltetrahydrofuran (2-MeTHF) solution. All complexes emit at 77 K with lifetimes on the order of 4 μs. A combined density functional theory (DFT) and time-dependent DFT (TDDFT) study reveals that the emission process is best described as a mixed metal-to-ligand/ligand-to-ligand charge transfer (MLCT/LLCT).
 Please wait while we load your content...
Please wait while we load your content...

