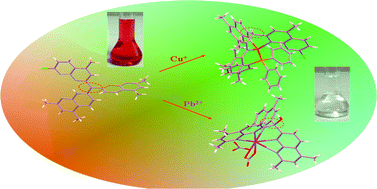
Two novel facial-capping tris-naphthyridyl compounds, 2-chloro-5-methyl-7-((2,4-dimethyl-1,8-naphthyridin-7(1H)-ylidene)(2,4-dimethyl-1,8-naphthyridin-7-yl))methyl-1,8-naphthyridine (L1) and 2-chloro-7-((2-methyl-1,8-naphthyridin-7(1H)-ylidene)(2-methyl-1,8-naphthyridin-7-yl))methyl-1,8-naphthyridine (L2), as well as their Cu(I) and Pb(II) complexes, [CuLa(PPh3)]BF4 (1) (PPh3 = triphenylphosphine, La = bis(2,4-dimethyl-1,8-naphthyridin-7-yl)(2-chloro-5-methyl-1,8-naphthyridin-7-yl)methane), [CuLb(PPh3)]BF4 (2) (Lb = bis(2-methyl-1,8-naphthyridin-7-yl)(2-chloro-1,8-naphthyridin-7-yl)methane), [Pb(OLa)(NO3)2] (3) (OLa = bis(2,4-dimethyl-1,8-naphthyridin-7-yl)(2-chloro-5-methyl-1,8-naphthyridin-7-yl)methanol) and [Pb(Lb)2][Pb(CH3OH)(NO3)4] (4), have been synthesized and characterized by X-ray diffraction analysis, MS, NMR and elemental analysis. The structural investigations revealed that the transfer of the H-atom at the central carbon to an adjacent naphthyridine-N atom affords L1 and L2 possessing large conjugated architectures, and the central carbon atoms adopt the sp2 hybridized bonding mode. The reversible hydrogen transfer and a geometric configuration conversion from sp2 to sp3 of the central carbon atom were observed when Pb(II) and Cu(I) were coordinated to L1 or L2. The molecular energy changes accompanying the hydrogen migration and titration of H+ to different receptor-N at L1 were calculated by density functional theory (DFT) at the SCRF-B3LYP/6-311++G(d,p) level in a CH2Cl2 solution, and the observed lowest-energy absorption and emission for L1 and L2 can be tentatively assigned to an intramolecular charge transfer (ICT) transition in nature.

 Please wait while we load your content...
Please wait while we load your content...

