
Recent advances on simulation and theory of hydrogen storage in metal–organic frameworks and covalent organic frameworks†
Abstract
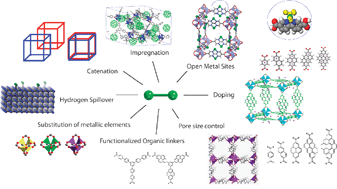
This critical review covers the application of computer simulations, including quantum calculations (ab initio and DFT), grand canonical Monte-Carlo simulations, and molecular dynamics simulations, to the burgeoning area of the hydrogen storage by metal–organic frameworks and covalent-organic frameworks. This review begins with an overview of the theoretical methods obtained from previous studies. Then strategies for the improvement of hydrogen storage in the porous materials are discussed in detail. The strategies include appropriate pore size, impregnation, catenation, open metal sites in metal
- This article is part of the themed collection: Metal Organic Frameworks
 Please wait while we load your content...
Please wait while we load your content...

