
Simulations of hydrogen, carbon dioxide, and small hydrocarbon sorption in a nitrogen-rich rht-metal–organic framework†

Abstract
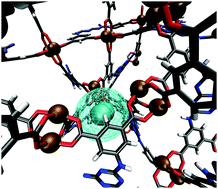
Grand canonical Monte Carlo (GCMC) simulations of gas sorption were performed in Cu-TDPAH, also known as rht-MOF-9, hereafter [1], a metal–organic framework (MOF) with rht topology consisting of Cu2+ ions coordinated to 2,5,8-tris(3,5-dicarboxyphenylamino)-1,3,4,6,7,9,9b-heptaazaphenalene (TDPAH) ligands. This MOF is notable for the presence of open-metal copper sites and high nitrogen content on the linkers. [1] Exhibits one of the highest experimental H2 uptakes at 77 K/1 atm within the extant rht-MOF family (ca. 2.72 wt%) and also has strong affinity for CO2 (5.83 mmol g−1 at 298 K/1 atm). Our simulations, which include explicit many-body polarization interactions, accurately modeled macroscopic thermodynamic properties (e.g., sorption isotherms and isosteric heats of adsorption (Qst)) as well as the binding sites for H2, CO2, CH4, C2H2, C2H4, and C2H6 in the MOF. Four different binding sites were observed through analysis of the radial distribution function (g(r)) about the two chemically distinct Cu2+ ions, simulated annealing calculations, and examination of the three-dimensional histogram showing the sites of occupancy: (1) at the Cu2+ ion facing toward the center of the linker (CuL), (2) at the Cu2+ ion facing away from the center of linker (CuC), (3) nestled between three [Cu2(O2CR)4] units in the corner of the truncated tetrahedral (T-Td) cage and (4) straddling the copper nuclei parallel to the axis of the Cu–Cu bond within the T-Td cage. The low-loading (initial) binding site in the MOF is highly sensitive to the partial charges of the Cu2+ ions that were used for parametrization. It was discovered that most sorbates prefer to sorb onto or near the Cu2+ ions that exhibit the greater partial positive charge (i.e., at site 1). The simulated H2 and CO2 sorption results obtained using a polarizable potential for the respective sorbates are in good agreement with the corresponding experimental data, especially near ambient pressure. Simulations of gas sorption were also performed in [1] using nonpolarizable potentials for the individual sorbates; these include potentials from the TraPPE force field for most sorbates.
 Please wait while we load your content...
Please wait while we load your content...

