
Long-range behavior of the transition dipole moments of heteronuclear dimers XY (X, Y = Li, Na, K, Rb) based on ab initio calculations†

Abstract
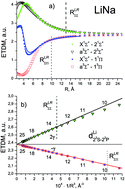
The ab initio electronic transition dipole moments (ETDMs) of heteronuclear dimers XY (X, Y = Li, Na, K, Rb) were calculated between the ground and excited states converging to the lowest three dissociation limits. The spin-allowed ETDMs were evaluated in a wide range of interatomic distances, R, by means of the quasi-relativistic electronic wave functions obtained by the multi-reference configuration interaction method. The inner-shell electrons (2 electrons for Li and Na atoms, and 10 and 28 for K and Rb, respectively) were described using the non-empirical shape-consistent effective core potentials. The l-independent core polarization potentials of each atom were used to take core-polarization and core-valence correlation effects into account. The long-range behavior of both singlet–singlet X1Σ+–(2,3)1Σ+;(1,2)1Π and triplet–triplet a3Σ+–(2,3)3Σ+;(1,2)3Π transition moments is perfectly fitted at large R-distance by the asymptotic formula of X. Chu and A. Dalgarno, Phys. Rev. A: At., Mol., Opt. Phys., 2002, 66, 024701: 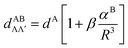 , where the coefficient β is equal to 2 and −1 for the Σ–Σ and Σ–Π transitions, respectively. The n2S–n2P transition moments, dA, and dynamic polarizabilites, αB, of the alkali atoms in the n2S state extracted from the present molecular calculations coincide with their empirical and ab initio counterparts to within a few percent.
, where the coefficient β is equal to 2 and −1 for the Σ–Σ and Σ–Π transitions, respectively. The n2S–n2P transition moments, dA, and dynamic polarizabilites, αB, of the alkali atoms in the n2S state extracted from the present molecular calculations coincide with their empirical and ab initio counterparts to within a few percent.
 Please wait while we load your content...
Please wait while we load your content...

