
Thermodynamic foundations of applications of ab initio methods for determination of the adsorbate equilibria: hydrogen at the GaN(0001) surface

Abstract
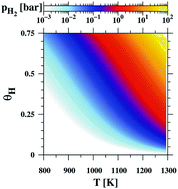
Thermodynamic foundations of ab initio modeling of vapor–solid and vapor–surface equilibria are introduced. The chemical potential change is divided into enthalpy and entropy terms. The enthalpy path passes through vapor–solid transition at zero temperature. The entropy path avoids the singular point at zero temperature passing a solid–vapor transition under normal conditions, where evaporation entropy is employed. In addition, the thermal changes are calculated. The chemical potential difference contribution of the following terms: vaporization enthalpy, vaporization entropy, the temperature-entropy related change, the thermal enthalpy change and mechanical pressure is obtained. The latter term is negligibly small for the pressure typical for epitaxy. The thermal enthalpy change is two orders smaller than the first three terms which have to be taken into account explicitly. The configurational vaporization entropy change is derived for adsorption processes. The same formulation is derived for vapor–surface equilibria using hydrogen at the GaN(0001) surface as an example. The critical factor is the dependence of the enthalpy of evaporation (desorption energy) on the pinning of the Fermi level bringing a drastic change of the value from 2.24 eV to −2.38 eV. In addition it is shown that entropic contributions considerable change the hydrogen equilibrium pressure over the GaN(0001) surface by several orders of magnitude. Thus a complete and exact formulation of vapor–solid and vapor–surface equilibria is presented.
 Please wait while we load your content...
Please wait while we load your content...

