
Theoretical study of one-electron-oxidized salen complexes of group 7 (Mn(iii), Tc(iii), and Re(iii)) and group 10 metals (Ni(ii), Pd(ii), and Pt(ii)) with the 3D-RISM-GMC-QDPT method: localized vs. delocalized ground and excited states in solution†

Abstract
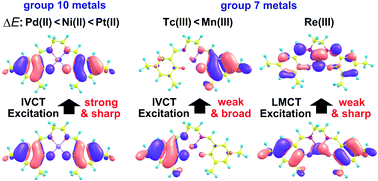
One-electron oxidized salen complexes of Mn(III) and Ni(II) were recently reported to be unique mixed-valence compounds. Their electronic structures are sensitive to the salen ligand and solvation. We systematically investigated a series of one-electron oxidized salen complexes of group 7 metals (Mn(III), Tc(III), and Re(III)) and their group 10 analogues (Ni(II), Pd(II), and Pt(II)) using the general multi-configuration reference quasi-degenerate perturbation theory (GMC-QDPT) which was combined with the three-dimensional reference interaction site model self-consistent field theory (3D-RISM-SCF) to incorporate the solvation effect. The calculated absorption spectra and electronic structures agree with the experimental observation. The one-electron oxidized salen complexes of group 10 metals with a symmetrical salen ligand have a delocalized electronic structure belonging to class III (Robin–Day classification) in weakly polar solvents. The tendency for taking a delocalized electronic structure increases in the order Pd(II) < Ni(II) < Pt(II). When the salen ligand is asymmetrical, the one-electron oxidized complexes have a localized electronic structure belonging to class II. The group 7 analogues of Mn(III) and Tc(III) have a localized electronic structure belonging to class II even in weakly polar solvents and even with a symmetrical salen ligand. However, the one-electron oxidized Re(III) complex has no mixed-valence nature because one-electron oxidation occurs on the Re center. Theoretical study shows that the solvation effect plays a crucial role in determining the mixed-valence character, class II or III, and thereby its incorporation in the calculation is indispensable for correctly describing geometries, electronic structures, and the inter-valence absorption spectra of these complexes. The d orbital energy is one of the most important factors for determining the localization/delocalization electronic structures in these complexes. Detailed discussion of these factors is presented.
 Please wait while we load your content...
Please wait while we load your content...

