
Theoretical investigations into the charge transfer properties of thiophene α-substituted naphthodithiophene diimides: excellent n-channel and ambipolar organic semiconductors†
 *a
*a
Abstract
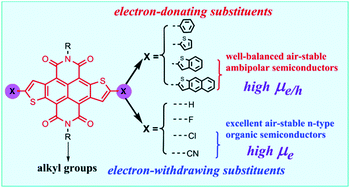
A theoretical study was carried out to investigate the electronic structures and the charge transport properties of a series of naphthodithiophene diimide (NDTI) thiophene α-substituted derivatives NDTI-X using density functional theory and classical Marcus charge transfer theory. This study deeply revealed the structure–property relationships by analyzing the intermolecular interactions in crystal structures of C8-NDTI and C8-NDTI-Cl thoroughly by using the Hirshfeld surface, QTAIM theories and symmetry-adapted perturbation theory (SAPT). Our results suggested that a 2-D brick-like π-stacking structure makes C8-NDTI-Cl a more excellent n-type semiconducting material with μmax-e of 2.554 cm2 V−1 s−1 than C8-NDTI with a herringbone-like slipped π-stacking motif. In addition, the calculated results showed that by modifying the thiophene α-positions of NDTI with electron-withdrawing substituents, –F, –Cl and –CN, low-lying LUMO energy levels and a high adiabatic electron affinity EA(a) can be obtained; while introducing electron-donating groups, benzene (–B), thiophene (–T), benzo[b]thiophene (–BT) and naphtha[2,3-b]thiophene (–NT), expanded the molecular π-conjugated backbone, and narrow band gaps, high EA(a) and small reorganization energies can be obtained. Theoretical simulations predict that NDTI-CN is an excellent air-stable n-type organic semiconducting material with an average electron mobility μe of up to 1.743 cm2 V−1 s−1. Owing to their high EA(a), moderate adiabatic ionization potential IP(a) as well as small hole and electron reorganization energies, NDTI-BT and NDTI-NT are two well-balanced air-stable ambipolar semiconducting materials. The theoretical average hole/electron mobilities are as high as 2.708/3.739 cm2 V−1 s−1 for C8-NDTI-NT and 1.597/2.350 cm2 V−1 s−1 for C8-NDTI-BT, respectively.
 Please wait while we load your content...
Please wait while we load your content...

