
Prediction of thermodynamically stable Li–B compounds at ambient pressure†
 a
Chao-Hao
Hu,
*bc
a
Chao-Hao
Hu,
*bc
Abstract
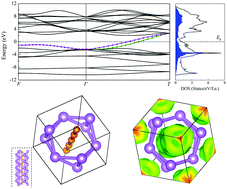
To clarify controversial structures and phase stability in the Li–B system, we predicted energetically favorable compounds and crystal structures of the Li–B binary system at ambient pressure, mainly including Li6B5, LiB2, and LiB3, from ab initio evolutionary structure simulations and further investigated physical properties of stable Li–B compounds using first-principles methods. Metallic Li6B5, predicted in our simulations, has trigonal symmetry with space group R32 and contains linear B chains, but its superconducting Tc is low according to the electron–phonon coupling calculations. Orthorhombic LiB2 (Pnma) and tetragonal LiB3 (P4/mbm) are zero-gap semiconductors; LiB2 is a Dirac semimetal, and both LiB2 and LiB3 are promising thermoelectric materials.
 Please wait while we load your content...
Please wait while we load your content...

