
Nonadiabatic photodynamics and UV absorption spectrum of all-trans-octatetraene†

Abstract
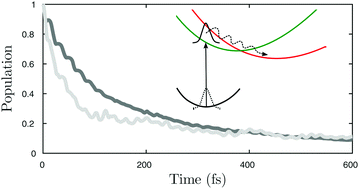
The short-time molecular quantum dynamics of all-trans-octatetraene after electronic excitation to the first bright valence state is theoretically investigated. A semiempirical approach of a multireference configuration interaction based on density functional theory, the so called hybrid DFT/MRCI, in both its original and redesigned formulations, is used for treating the electronic part of the problem. The nuclear kinetic part is defined with the help of symmetry-adapted internal coordinates also suitable for a large amplitude displacement. By incorporating ten in-plane and two out-of-plane nuclear degrees of freedom in the underlying Hamiltonian, the results of the time evolution of the excited wave packet are discussed. We show that the population transfer between the two coupled low-lying states in all-trans-octatetraene occurs in a 100–200 fs time regime. The calculated UV absorption spectra describe the main vibronic features correctly except for the band associated with the single-bond stretching motion which lacks intensity. The possible products of the photoisomerization in terms of symmetry-adapted coordinates are also discussed.
 Please wait while we load your content...
Please wait while we load your content...

