
Modelling the local atomic structure of molybdenum in nuclear waste glasses with ab initio molecular dynamics simulations†
Abstract
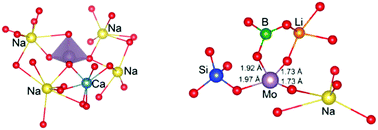
The nature of chemical bonding of molybdenum in high level nuclear waste glasses has been elucidated by ab initio molecular dynamics simulations. Two compositions, (SiO2)57.5–(B2O3)10–(Na2O)15–(CaO)15–(MoO3)2.5 and (SiO2)57.3–(B2O3)20–(Na2O)6.8–(Li2O)13.4–(MoO3)2.5, were considered in order to investigate the effect of ionic and covalent components on the glass structure and the formation of the crystallisation precursors (Na2MoO4 and CaMoO4). The coordination environments of Mo cations and the corresponding bond lengths calculated from our model are in excellent agreement with experimental observations. The analysis of the first coordination shell reveals two different types of molybdenum host matrix bonds in the lithium sodium borosilicate glass. Based on the structural data and the bond valence model, we demonstrate that the Mo cation can be found in a redox state and the molybdate tetrahedron can be connected with the borosilicate network in a way that inhibits the formation of crystalline molybdates. These results significantly extend our understanding of bonding in Mo-containing nuclear waste glasses and demonstrate that tailoring the glass composition to specific heavy metal constituents can facilitate incorporation of heavy metals at high concentrations.
 Please wait while we load your content...
Please wait while we load your content...

