
Vibrational spectroscopy of Methyl benzoate†
Abstract
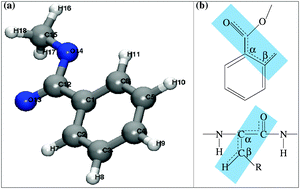
Methyl benzoate is studied as a model compound for the development of new IR pulse schemes with possible applicability to biomolecules. Anharmonic vibrational modes of Methyl benzoate are calculated on different level (MP2, SCS, CCSD(T) with varying basis sets) ab initio PESs using the vibrational self-consistent field (VSCF) method and its correlation corrected extensions. Dual level schemes, combining different quantum chemical methods for diagonal and coupling potentials, are systematically studied and applied successfully to reduce the computational cost. Isotopic substitution of β-hydrogen by deuterium is studied to obtain a better understanding of the molecular vibrational coupling topology.
 Please wait while we load your content...
Please wait while we load your content...

