
Molecular dynamics simulations of proton-ordered water confined in low-diameter carbon nanotubes

Abstract
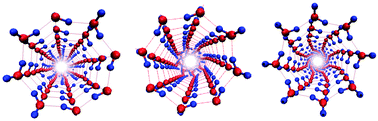
The present work deals with molecular dynamics simulations of water confined in single-walled carbon nanotubes (CNTs), with emphasis on the proton-ordering of water and its polarization. First, the water occupancy of open-ended armchair and zigzag CNTs immersed in water under ambient NPT conditions is calculated for various water models, and for varying Lennard-Jones parameters of the water–carbon interaction. As a function of the CNT diameter, the water density displays several oscillations before converging to the bulk value. Based on these results, the water structures encapsulated in 10 nm long armchair CNTs (n,n) with 5 ≤ n ≤ 10, are investigated under NVT conditions. Inside the smallest nanotubes (n = 5, 6) highly ferroelectric (FE), quasi-one-dimensional water chains are found while inside the other CNTs water molecules assemble into single-walled ice nanotubes (INTs). There are several, near-degenerate minimum energy INT structures: single helical structures were found for 7 ≤ n ≤ 10, in all cases in FE arrangement. In addition, a double helical INT structure was found for n = 8 with an even higher polarization. Prism-like structures were found only for 8 ≤ n ≤ 10 with various FE, ferrielectric (FI), and antiferroelectric (AF, n = 9, 10) proton ordering. The coexistence of the nearly iso-energetic FE, FI, and AF INT structures separated by high barriers renders the molecular dynamics highly metastable, typically with nanosecond timescales at room temperature. Hence, the replica exchange simulation method is used to obtain populations of different INT states at finite temperatures. Many of the FE INT structures confined in low-diameter CNTs are still prevalent at room temperature. Both helix–helix and helix–prism structural transitions are detected which can be either continuous (around 470 K for n = 8) or discontinuous (at 218 K for n = 9). Also melting-like transitions are found in which the INT structures are disrupted leading to a loss of FE or FI ordering of the water orientations. Also these transitions can be either smooth (for n = 7, 8) or abrupt, first-order transitions, at T = 362 K for n = 9 and at T = 285 K for n = 10.
 Please wait while we load your content...
Please wait while we load your content...

