
Pushing the limits in accurate vibrational structure calculations: anharmonic frequencies of lithium fluoride clusters (LiF)n, n = 2–10†
Abstract
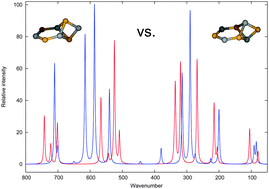
The vibrational spectra of a series of small lithium fluoride clusters, i.e. (LiF)n, n = 2–10, were studied by vibrational configuration interaction (VCI) calculations relying on potential energy surfaces including three-mode coupling terms and being obtained from explicitly correlated local coupled cluster calculations. Due to the account for anharmonicity effects, the simulated spectra allow for a direct comparison with experimental data and may thus help to identify clusters in the experiments. Even structurally closely related clusters can clearly be distinguished by infrared spectroscopy. The largest system in this study required more than 1000 basis functions in the electronic structure calculations and more than 107 configurations in the vibrational structure calculations and became computationally feasible only due to a combination of different approximations and highly parallelized algorithms.
 Please wait while we load your content...
Please wait while we load your content...

