
Performance of conventional and dispersion-corrected density-functional theory methods for hydrogen bonding interaction energies†
Abstract
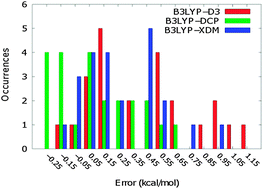
The approximate CCSD(T)/CBS binding energies for the set of 23 hydrogen-bonded dimers (HB23) of the S66 set reported by Řezáč et al. (J. Chem. Theory Comput. 2011, 7, 2427–2438) were expected to be under-estimated based on the known under-binding tendency of the counterpoise correction combined with small basis sets. In this work, we present binding energies for the HB23 set of dimers obtained using a composite approach recently described by Mackie and DiLabio (J. Chem. Phys. 2011, 135, 134318) that averages the counterpoise- and non-counterpoise-corrected energies, while utilizing standard approaches to obtain CCSD(T)/CBS-type energies. The binding energies for the HB23 set are revised upward by an average of 0.12 kcal mol−1 and as much as 0.35 kcal mol−1. We use these improved benchmark-level binding energies to evaluate the ability of pure, hybrid, long-range-corrected, and dispersion-corrected density-functional theory (DFT) methods to accurately predict the binding energies of hydrogen-bonded dimers. We find that, in general, the inclusion of dispersion into the DFT approach is required in order to obtain reasonable results for the HB23 set. We find that the dispersion-corrected DFT methods we tested produce results of variable quality, as measured by mean absolute deviation relative to the revised reference values we computed: B97D, 0.57 kcal mol−1; B3LYP-D3, 0.44 kcal mol−1; ωB97XD, 0.25 kcal mol−1; LC-ωPBE-D3, 0.24 kcal mol−1; M06-2X, 0.21 kcal mol−1; B3LYP-DCP, 0.23 kcal mol−1; B971-XDM, 0.18 kcal mol−1.
 Please wait while we load your content...
Please wait while we load your content...

